


Содержание
Перейти к:
https://doi.org/10.35754/0234-5730-2021-66-4-580-592
Перейти к:
Введение. Образование клеток крови у здорового человека обеспечивается поликлональным кроветворением. С возрастом в периферической крови обнаруживаются большие клоны, происходящие из одной клеткипредшественницы, что является проявлением клонального кроветворения. В некоторых случаях у лиц с клональным кроветворением развиваются гематологические заболевания.
Цель обзора — описать и обобщить современные данные о связи клонального кроветворения с гематологическими заболеваниями.
Основные сведения. Описывается история выявления клонального кроветворения, основные его свойства, наиболее частые мутации в кроветворных клонах, связанные с риском развития миелодиспластического синдрома и острого миелоидного лейкоза. Обсуждается возможный патогенез опухолевой трансформации.
Петинати Н.А., Дризе Н.И. Клональное кроветворение и его роль в развитии гематологических заболеваний. Гематология и трансфузиология. 2021;66(4):580-592. https://doi.org/10.35754/0234-5730-2021-66-4-580-592
Petinati N.A., Drize N.J. Clonal hematopoiesis and its role in the development of hematological diseases. Russian journal of hematology and transfusiology. 2021;66(4):580-592. (In Russ.) https://doi.org/10.35754/0234-5730-2021-66-4-580-592
Нормальное кроветворение поддерживается множеством долго и коротко живущих, сменяющих друг друга клонов кроветворных клеток. Костный мозг — одна из самых быстро пролиферирующих тканей. В организме человека производится примерно 1 триллион зрелых клеток кровикаждыйдень [1]. Воснове системы кроветворения лежат стволовые кроветворные клетки (СКК), способные дифференцироваться во все зрелые клетки крови [2],[3]. Потомство одной СКК представляет собой клон [4]. При устойчивом кроветворении система поликлональна, т. е. множество СКК одновременно участвует в поддержании нормальных показателей клеток периферической крови [5],[6],[7]. Менее 1,3 миллиона СКК поддерживают образование зрелых клеток периферической крови за жизнь человека [8]. Клональное кроветворение (КК) представляет собой непропорциональное увеличение числа потомков только нескольких СКК, несущих определенные мутации, по отношению к множеству остальных клонов. КК — это биологическое состояние, а не заболевание. При лейкозах и лимфомах происходит размножение клональных клеток, которые не способны дифференцироваться и ингибируют нормальное кроветворение. Неопластические клоны могут происходить из плюрипотентных СКК или более коммитированных миелоидных или лимфоидных клеток-предшественниц. Фенотип образовавшейся злокачественной опухоли обусловлен приобретенными генетическими и эпигенетическими изменениями, а также уровнем дифференцировки клетки, из которой она происходит [9].
Цель настоящего обзора — описать и обобщить современные данные о связи клонального кроветворения с гематологическими заболеваниями.
КК определяется несбалансированным вкладом одной СКК в периферическую кровь и может возникнуть в результате нейтрального дрейфа или направленного отбора. При нейтральном дрейфе все клоны имеют одинаковый шанс внести свой вклад в пул самообновляющихся СКК, но случайные процессы, такие как истощение пула стволовых клеток, приводят к превалированию созревания одних клонов над другими. При положительном отборе соматические изменения опосредуют преимущество избирательного роста определенных клонов СКК относительно других, что приводит к клональному доминированию. КК — это количественная характеристика, очень похожая на артериальное давление и рост. Хотя КК было определено как бинарная характеристика (есть/нет), истинная природа КК описывается клональным разнообразием системы крови. Генетическое разнообразие определяется количеством СКК у одного человека и их относительным вкладом в пул зрелых клеток крови. Это генетическое разнообразие может изменяться с возрастом и по другим причинам, таким как апластическая анемия, цитостатическое воздействие и т. д. [10]. У лиц с КК чаще всего выявляются нарушения в генах ДНК-метилтрансферазы 3 альфа (DNMT3A), регулятора транскрипции ASXL1, транслокации десять-одиннадцать-2 (TET2), а также янус-киназы 2 (JAK2),фактора сплайсинга 3 В субъединицы 1 (SF3B1), фактора сплайсинга серина/аргинина обогащенного 2 (SRSF2) и опухолевого белка р53 (TP53). Мутации в генах DNMT3A, ASXL1, TET2 с возрастом приводят к клональной экспансии (увеличению количества клеток одного или нескольких клонов). Такая ситуация в кроветворной системе совместима с нормальными показателями крови, но предрасполагает к онкогематологическим и сердечно-сосудистым заболеваниям [11],[12],[13],[14]. Мутации, характерные для КК, были выявлены у большинства пожилых людей [15]. Частота обнаружения мутаций намного больше, чем частота онкогематологических заболеваний в общей популяции. КК может быть предшествующим событием в развитии лейкозов [16]. Мутации в генах JAK2, SF3B1, SRSF2 и TP53 ранее были определены как драйверные при различных миелоидных новообразованиях, включая миелопролиферативные заболевания (МПЗ), миелодиспластический синдром (МДС) и острый миелоидный лейкоз (ОМЛ) [17]. В настоящее время интенсивно исследуются клеточные и внешние факторы, связанные со злокачественной эволюцией КК, и можно надеяться, что результаты этих работ приведут к разработке стратегий раннего определения, мониторинга и, в итоге, предупреждения заболеваний. История изучения КК началась в середине прошлого века.
Впервые КК было выявлено у женщин на основе принципа случайной инактивации Х-хромосомы (ИXХ) [18]. ИИX происходит в клетках самок млекопитающих для того, чтобы с двух копий X-хромосом не образовывалось вдвое больше продуктов соответствующих генов, чем у самцов. На ранних стадиях эмбриогенеза выбор X-хромосомы, которая будет инактивирована, случаен. Этот выбор, сделанный однажды, сохраняется пожизненно в этой клетке и во всех ее потомках. Таким образом, в половине клеток организма функционирует одна из Х-хромосом, а во второй половине — другая. Перекос в этом распределении свидетельствует о наличии доминантных клонов. У взрослой женщины присутствует мозаицизм клеток с активной Х-хромосомой отца или матери. На Х-хромосоме расположен ген фермента глюкозо-6-фосфат дегидрогеназы. Этот фермент существует в двух формах, которые различаются по подвижности на электрофорезе. У женщин, гетерозиготных по этому ферменту, легко определить соотношение активности отцовской и материнской Х-хромосомы. Данные 10-летнего анализа ИХХ у женщин разного возраста, гетерозиготных по этому гену, в разных тканях и при различных гематологических заболеваниях позволили F. Fialkow сделать вывод о клональном развитии опухолей человека [19]. Этот факт в дальнейшем был подтвержден с помощью более надежных и информативных методов исследования [20]. Однако доказательства того, что генетический компонент, связанный с Х-хромосомой, определяется ИХХ, были впервые получены только спустя 20 лет [21]. J. Abkowitz и соавт. [22] проанализировали кроветворные клетки стареющих самок кошек породы сафари. Эта порода была выведена путем скрещивания кошек жоффруа с домашними кошками. У самок таких гибридов в половых клетках не может происходить рекомбинация между двумя родительскими X-хромосомами, соответственно активна глюкозо-6-фосфат дегидрогеназа жоффруа или домашних кошек. Авторы обнаружили, что ИХХ была случайной у молодых кошек, но у 67 % кошек старшего возраста наблюдался перекос, всегда в сторону Geoffroy X-хромосомы. Этот факт указывал на отбор гемизиготных клеток и не мог быть случайным. Следовательно, этот перекос был связан с преобладанием отдельных клонов, то есть переходом к клональному кроветворению. Эта работа послужила новым толчком к изучению ИХХ у людей. Исследовали соотношение ИХХ в тканях 2292 женщин французско-канадского происхождения без каких-либо известных гематологических заболеваний, в возрасте от 37 до 101 года, из них 1734 женщины принадлежали к 311 семьям и 558 не были связаны между собой родственными узами. Перекос в ИХХ чаще встречался в миелоидных клетках, чем в Т-лимфоцитах или клетках буккального эпителия и зависел от возраста женщины. Авторы предположили, что перекос мог быть вызван разными факторами, в том числе мутациями. С помощью полногеномного секвенирования экзома 3 пожилых женщин с перекосом ИХХ в миелоидных клетках и поликлональными Т-клетками идентифицировали соматические мутации в генах TET2, DNMT3A и двенадцатого члена семейства носителей растворенных веществ (SLC39A12) у одной из них. Включение в анализ представительниц различных возрастных групп позволило выявить нонсенс и миссенс соматические мутации и сдвиг рамки считывания в гене TET2 в ДНК только из миелоцитов. При этом в лимфоцитах или эпителиальных клетках перекоса не было. Эти изменения определялись у 10 (5,6 %) из 179 женщин с перекосом в ИХХ и ни у одной из 105 пожилых женщин без перекоса в ИХХ, а также ни у одной из 96 более молодых женщин с перекосом в ИХХ. У женщин, имевших мутации в гене TET2, показатели крови не отличались от показателей женщин соответствующего возраста, не имевших мутаций в этом гене. Это исследование показало, что приобретенная мутация в гене, ассоциированном с миелоидными гемобластозами — TET2, зависит от возраста и не вызывает патологии кроветворения [22].
При исследовании однонуклеотидных полиморфизмов у более чем 50 000 человек клональный мозаицизм описан у 2–3 % людей пожилого возраста и у 0,5 % людей моложе 50 лет. Показано, что наличие клонального мозаицизма ассоциируется с 10-кратным увеличением риска развития онкогематологических заболеваний [23]. Развитие новых методов исследования, в том числе полногеномного секвенирования, привело к тому, что в 2014 г. сразу три группы исследователей опубликовали данные об экзомах (последовательности, относящиеся к кодирующей части генома) ДНК больших групп больных и выявили возрастные мутации в драйверных генах [11],[13],[24]. Описанные группы включали 2728 человек солидными опухолями, 12 380 человек и 17 182 человека разного возраста без гематологических заболеваний. Во всех группах в образцах периферической крови были выявлены мутации в сходном наборе генов, при этом встречаемость этих мутаций зависела от возраста. Было идентифицировано более 70 различных генов, несущих мутации. Наиболее часто мутировавшими были гены, ассоциированные с КК — DNMT3A, TET2 и ASXL1. Результаты дальнейшего наблюдения и обследования, доступные для двух из этих исследований, показали, что у людей с выявленным КК риск развития гемобластоза был повышен в 10 раз [11],[13].
При клональной экспансии СКК отсутствуют анатомические ограничения, характерные для других тканей, поскольку эти клетки и их потомство циркулируют в крови в очень большом количестве [25]. Следовательно, при КК возрастает вероятность не только злокачественной трансформации клона (чаще миелоидной направленности, реже — лимфоидной), вызванной приобретением вторичных мутаций, но также развития сердечно-сосудистой патологии и, скорее всего, других заболеваний, на которые влияет измененнаяфункция мутантных клеток крови. Понимание того, как возникает КК и как происходит клональный отбор и экспансия клонов, имеет большое значение для представлений о биологии старения организма и развития различных заболеваний, а также для разработки стратегии предотвращения клинических последствий, связанных с КК. Возникающее у пожилых людей КК относится к клональной экспансии и не связано с типом мутаций [26]. С помощью метода полногеномного секвенирования была подтверждена широкая распространенность мутаций в драйверных генах. Мутаций в генах-кандидатах, не являющихся драйверами, оказалось еще больше [27]. При использовании еще более чувствительного по сравнению с полногеномным секвенированием метода, ориентированного непосредственно на гены, был проведен анализ 2530 нормальных людей в возрасте от 55 до 101 года [28]. Это исследование выявило высокую встречаемость мутаций в генах DNMT3A или TET2, играющих центральную роль в метилировании и деметилировании ДНК. Не было обнаружено существенных различий в анализах крови людей с мутантными клонами и без них, за исключением тенденции к уменьшению количества полиморфноядерных клеток у людей с мутацией гена TET2. Было показано, что мутации в гене DNMT3A происходили в СКК, тогда как мутации в гене TET2 встречались главным образом в миелоидных клетках, редко — в B-клетках, т. е. происходили на разных уровнях кроветворной иерархии [28]. Во всех представленных работах КК наблюдалось у пожилых людей.
Чем отличаются параметры кроветворной системы у молодых и пожилых? Существует множество различий между молодыми и старыми СКК [29]. Старение СКК ассоциировано с увеличением их количества [30], уменьшением функциональности индивидуальных СКК [31], уменьшением полярности клеток и нарастанием количества повреждений ДНК [32], уменьшением длины теломер [33] и снижением уровня аутофагии [34]. При этом все основные функции СКК сохраняются. Все эти изменения преимущественно внутренние. Эпигенетические возрастные изменения включают метилирование ДНК и ремодуляцию хроматина [35]. Некоторые из этих изменений представляют собой признаки неизбежной гибели кроветворных клеток, а другие связаны с адаптационными механизмами кроветворной системы [16]. В нормальных СКК соматические мутации ассоциированы с возрастом и представляют переход азотистых оснований NрCрG (цитозин-гуанин) в тринуклеотидных последовательностях в результате спонтанного дезаминирования 5’-метилцитозина до СрТ тимина (цитозин-тимин). Такие замены оснований накапливаются в геномах СКК со скоростью 14 мутаций в год, включая мутации в экзонах генов со скоростью 1 замена каждые 10 лет [36]. Таким образом, стабильный поликлональный гемопоэз отражает сбалансированный состав множества отдельных клонов, каждый из которых теоретически определяется по соматическим генетическим отличиям [37].
На протяжении процесса старения все меньше и меньше СКК вносят «вклад» в кроветворение, хотя на общее количество зрелых клеток крови это сильно не влияет. При исследовании клеток крови 115 летней женщины с помощью глубокого полногеномного секвенирования было обнаружено 450 соматических мутаций, в основном, в некодирующей области АТ-богатых участков генома. Эти мутации не привели к развитию патологии. Распределение частот вариантных аллелей (ЧВА) этих мутаций позволило предположить, что большинство лейкоцитов периферической крови были потомками всего двух родственных клонов СКК. Кроме того, длина теломер в клетках периферической крови была значительно короче, чем в клетках других тканей. Авторы этой работы считают, что ограничения продолжительности жизни или количества СКК, способных к пролиферации и дифференцировке, а не эффекты соматических мутаций, могут привести к клональной эволюции кроветворения в экстремальном возрасте [38].
Для исследования влияния наследственных факторов на КК было проведено глубокое направленное секвенирование ДНК из клеток крови 52 однояйцевых (монозиготных) и 27 разнояйцевых (дизиготных) пар близнецов в возрасте от 70 до 99 лет. С помощью этого высокочувствительного подхода в 62 % случаев было идентифицировано КК (ЧВА > 0,5 %). Частота КК была одинаковой у монозиготных и дизиготных пар близнецов и не отличалась от общей популяции. Были идентифицированы две монозиготные пары, в которых оба близнеца несли идентичные редкие соматические мутации, что указывало на общее происхождение их клеток. У трех монозиготных пар близнецов в образцах крови, взятых с разницей в 4–5 лет, обнаружили выраженную изменчивость динамики развития клонов, несущих мутации в одном и том же драйверном гене. Результаты этого исследования показали, что унаследованный геном не оказывал доминирующего влияния на КК у взрослых. Вместе с тем мутации при КК могут быть приобретены внутриутробно [39]. В популяционном исследовании анализировали 594 близнеца в возрасте от 73 до 94 лет, период наблюдения составил более 20 лет. Были выявлены мутации у 214 (36 %) близнецов, 20 пар близнецов имели мутации в одних и тех же генах, при этом одинаковые мутации наблюдались только у двух пар близнецов. Не было существенной разницы в мутациях конкретных генов, подгрупп или клонов кроветворных клеток между монозиготными и дизиготными близнецами. Результаты этого исследования подтверждают, что КК определяется не наследственными факторами [40]. Для прямого измерения связи КК с семейными факторами сравнили продолжительность жизни близнецов в парах, где у одного выявлялись мутации, а у другого — нет. Всего в исследование было включено 127 пар близнецов и в 48 % случаев близнец с мутациями умер первым. В этом исследовании также не обнаружено влияния наследственных факторов на КК [41].
В обоих исследованиях были обнаружены внутриутробные мутации, характерные для КК, некоторые из которых рассматривают как предшествующие лейкозу, причем оба близнеца несли одну и ту же мутацию. В отдельных случаях только по прошествии многих лет развился фенотип КК, несмотря на существование мутации в СКК в течение, по крайней мере, 50 лет жизни. Приобретение мутаций, предшествующих лейкозу, внутриутробно может быть даже более распространенным, но оставаться не выявленным изза отсутствия фенотипических изменений. Результаты исследований близнецов позволяют предположить, что с возрастом КК более подвержено внешним факторам, поэтому важен поиск факторов, влияющих на КК [42].
В зависимости от типа и количества соматических мутаций КК можно разделить на КК с неопределенным потенциалом и КК с онкогенным потенциалом. В то время как при КК с неопределенным потенциалом мутации сами по себе обычно создают молекулярный фон неопластического процесса, мутации КК с онкогенным потенциалом связаны с заболеваниями или даже специфическими поражениями, которые вызывают дифференциацию и/или пролиферацию опухолевых клеток. Со временем приобретение дополнительных генетических нарушений приводит к трансформации КК в ОМЛ. В таблице 1 представлена вероятность трансформации КК в гематологические заболевания в зависимости от гена, несущего перестройку.
Таблица 1. Примеры мутаций, которые были описаны в контексте КК с неопределенным потенциалом или связанных с возрастом и встречающихся у больных различными гематологическими заболеваниями [43]
Table 1. Examples of mutations that have been described in the context of CH of indeterminate potential or age-related and determined in patients with various hematological malignancies [43]
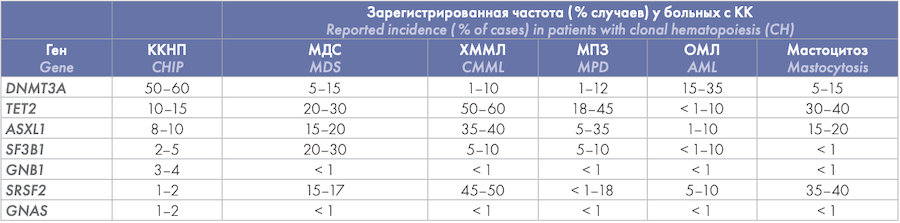
Примечание. ККНП — КК с неопределенным потенциалом, ХММЛ — хронический мономиелоцитарный лейкоз, МПЗ — миелопролиферативное заболевание, ОМЛ — острый миелоидный лейкоз.
Note. CHIP — clonal hematopoiesis of indeterminate potential, CMML — chronic monomyelocytic leukemia, MPD — myeloproliferative disease, AML — acute myeloid leukemia.
КК с точечными мутациями или структурными вариациями, такими как изменение числа копий гена, приводит к 10-кратному увеличению риска развития злокачественных новообразований. Хотя абсолютный риск злокачественных заболеваний системы крови низкий, некоторые варианты КК, включая присутствие мутаций в гене TP53 или генах сплайсосомы, при ЧВА больше 10 %, а также наличие множественных мутаций и измененные показатели эритроцитов могут повышать риск злокачественной трансформации. КК при цитопении ассоциировано с очень высоким риском прогрессии в миелоидную злокачественную опухоль, и такие больные требуют пристального наблюдения. Токсическое воздействие на гены химиотерапии или лучевой терапии и хронического воспаления может увеличить риск трансформации доминирующих клонов в миелоидную злокачественную опухоль [44]. СКК, которые приобретают соматические мутации, дающие конкурентное преимущество относительно их нормальных аналогов, могут образовывать большие клоны, что приводит к КК. Мутации в генах, часто измененных при КК, также периодически выявляются при миелоидных злокачественных новообразованиях. Таким образом, КК может приводить к развитию злокачественного кроветворения. Отношение рисков развития гематологических злокачественных новообразований у больных с КК было 11,1 [11],[27].
При изучении однонуклеотидных полиморфизмов для определения КК в течение длительного времени хромосомные аномалии многократно обнаружили у 149 из 8562 человек (1,7 %) [23]. В другом исследовании использовали массивы однонуклеотидных полиморфизмов для анализа 151 202 участников биобанка Великобритании, позволяющие обнаруживать хромосомные аномалии всего в 1 % фракции клеток. Используя этот подход, идентифицировали КК в 7484 (4,9 %) случаях [45]. По сравнению с людьми без КК риск злокачественного кроветворения у лиц с КК был увеличен в 10 раз с поправкой на возраст [45]. В других исследованиях наблюдали связь между обнаружением КК и развитием опухолевых заболеваний крови. Наиболее сильная связь КК была выявлена с развитием хронического лимфолейкоза (ХЛЛ) [44].
Мутации, обнаруженные у больных миелоидными злокачественными новообразованиями, могут быть разделены на несколько отдельных групп в зависимости от их воздействия на клеточные компоненты или функциональные пути [46]. Связь между КК и развитием ОМЛ была выявлена в двух исследованиях, в которых сравнили образцы периферической крови, полученной от здоровых людей, накопленных за несколько лет до установления диагноза ОМЛ, с соответствующими образцами периферической крови, полученной от лиц, у которых никогда не развивался ОМЛ [47], [48]. Р. Desai и соавт. [47] анализировали данные крупного проспективного исследования женского здоровья. Они оценили 212 женщин, у которых развился ОМЛ, и 212 женщин соответствующего возраста, у которых ОМЛ не развился, и обнаружили связь между мутациями, характерными для КК, с ЧВА > 1 % и увеличением вероятности развития ОМЛ (отношение шансов (OШ) — 4,86, 95 % доверительный интервал (ДИ): 3,07–7,77). S. Abelson и соавт. [48] проанализировали 95 человек, у которых развился ОМЛ, и 414 людей, которые не заболели лейкозом. Геномные последовательности для анализа были получены из базы данных крупного исследования связи развития онкологических заболеваний с питанием (EPIC). Было показано, что существует повышенный риск развития ОМЛ у лиц с мутациями КК при ЧВА более 0,5 %. При анализе данных исследования профессионального здоровья (Nurses’ Health Study and Health Professionals Follow-Up Study), проведенном A. L. Young и соавт. [49], у 35 наблюдаемых, у которых развился ОМЛ при сравнении с теми, у кого ОМЛ не развился, вероятность развития лейкоза была повышена при ЧВА ≥ 1 % (ОШ — 5,4, 95 % ДИ: 1,8–16,6; р = 0,003). Ранее для определения КК неопределенного потенциала применялся порог ЧВА 2 % [50]. Используя прицельное глубокое секвенирование, в исследованиях P. Desai и соавт. [47] и S. Abelson и соавт. [48] показано, что у 70 % больных, у которых было выявлено КК, развился ОМЛ, в то время как в контрольной группе, в которой КК не выявлялось, ОМЛ был выявлен лишь в 30 % случаев. Средний латентный период составил 8,9 и 6,8 года, соответственно, для двух исследований [47],[48]. Наиболее распространенные мутации, выявленные у больных, у которых развился ОМЛ, обнаружены в генах: DNMT3A, TET2, TP53, изоцитрат дегидрогеназы 1 (IDH1), и 2 (IDH2), JAK2 и в генах сплайсосомы. В другом исследовании, изучавшем КК, показано, что эти мутации могут быть и у здоровых людей пожилого возраста [51].
Наличие КК повышает риск развития как первичного, так и вторичного ОМЛ. Мутации в генах TP53, IDH1/2, DNMT3A, TET2 и сплайсосомы при КК ассоциированы с первичным ОМЛ, в то время как мутации в генах магний-зависимой протеинфосфатазы 1 (PPM1D) и TP53 повышают риск развития вторичного МДС/ОМЛ.
Определены гены, наиболее часто повреждаемые при миелоидных неоплазиях (табл. 2). Мутации большинства из этих генов обнаружены при КК и считаются драйверными мутациями при развитии неоплазий [52]. У пожилых людей при КК наиболее распространены мутации в генах DNMT3A и TET2 — на них приходится 45–55 % и 10–30 % всех мутаций соответственно. DNMT3A и TET2 это гены, участвующие в регуляции метилирования и деметилирования ДНК [53].
Таблица 2. Мутации в генах, часто наблюдаемые при КК и ОМЛ
Table 2. Gene mutations commonly detected in CH and AML
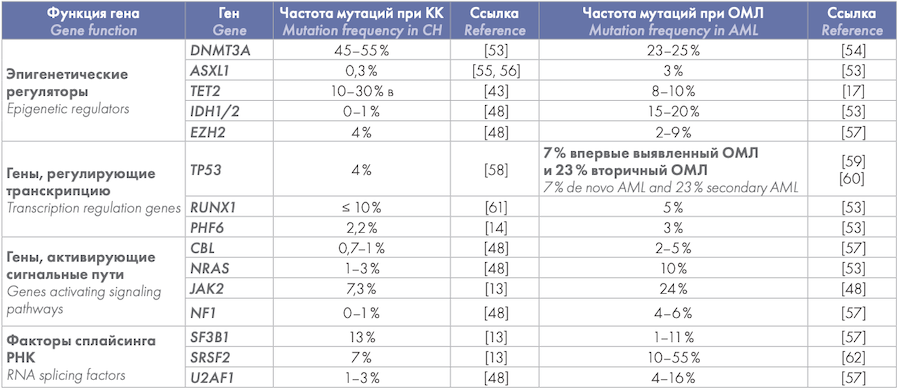
У больных ОМЛ эти мутации составляют 23–25 % и 8–10 %, соответственно [17],[54]. Мутации в гене TET2, ведущие к потере функции, обнаружены у 15– 25 % больных различными миелоидными новообразованиями [63]. У больных ОМЛ частота мутаций в гене TET2 увеличивается с возрастом [64], и по сравнению с нормальными образцами костного мозга при ОМЛ с мутациями в гене TET2 обнаруживается специфическая ДНК с фенотипом гиперметилирования, приводящего к аберрантной экспрессии этого гена [65]. На функциональном уровне снижение экспрессии TET2 связано с клональной экспансией СКК, особенно в ответ на воспалительный стресс [66].
ASXL1 — еще один рекуррентно мутирующий ген при КК, который также кодирует белок, связанный с эпигенетической модификацией. В отличие от DNMT3A и TET2, которые непосредственно модифицируют ДНК, ASXL1 входит в так называемую поликомб-группу, которая регулирует транскрипцию посредством модификации гистонов [67]. Мутации в гене ASXL1 встречаются при различных миелоидных злокачественных новообразованиях, большинство обычно представляют собой варианты со сдвигом рамки считывания или нонсенс-мутациями, которые ассоциируются с плохим прогнозом при ОМЛ [64], МДС [68] и ХММЛ [69].
Мутации в гене IDH1/2 ассоциированы со значительным риском прогрессии ОМЛ в исследованной популяции, эти мутации выявляются у 15–20 % больных впервые диагностированным ОМЛ [53]. В исследованиях P. Desai и соавт. [14] и А. L. Young и соавт. [51] у всех людей с мутациями IDH1/2, выявленными в момент отбора исходных образцов, в итоге развился ОМЛ.
Мутации в гене TP53 относят к другой подгруппе мутаций КК, при которых высок риск развития ОМЛ. Мутации в гене TP53 составляют 4 % мутаций КК в популяции [58] и присутствуют у 7 % больных впервые диагностированным ОМЛ и 23 % больных вторичными ОМЛ, возникшими после предшествующей химиотерапии [59],[60],[70],[71],[72].
Сплайсосомные мутации: SRSF2, вспомогательного фактор 1 малой ядерной РНК U2 (U2AF1), SF3B1, CCCH-тип цинкового пальца, мотив связывания РНК и богатый серином/аргинином 2 (ZRSR2) составляют 5 % мутации при КК [58], но очень часто встречаются при МДС (50 %) и ОМЛ (первичный ОМЛ — 10 %, вторичный ОМЛ — 10–55 %) [62].
Мутации JAK2-V617F обычно присутствуют при МПЗ [73] и ассоциированы с повышенным риском тромботических осложнений даже у больных, у которых нет МПЗ. В исследованиях S. Abelson и соавт. [48] и P. Desai и соавт. [47] показано, что наличие JAK2-V617F мутации при КК связано с повышенным риском ОМЛ (ОШ — 6,1, 95 % ДИ: 1,2–61,1; p = 0,03).
Мутации в гене PHD цинкового пальца 6 (PHF6) и фактора транскрипции 1, родственного runt (RUNX1) редко обнаруживают при КК (в 3 из 424 случаев в исследовании P. Desai и соавт. [47] и в 8 из 105 случаев в работе A. L. Young и соавт. [49]), но заслуживают обсуждения, поскольку у всех людей с мутациями в гене PHF6 в вышеупомянутых исследованиях развился ОМЛ, а в одном из исследований была показана связь с быстрым развитием ОМЛ в течение 5 лет [14].
Неизвестно, возникают ли множественные мутации при КК в разных клонах, или они возникают в результате последовательного появления новых мутаций в одиночном клоне. Не исключено, что риск развития ОМЛ может быть различным в зависимости от происхождения этих мутаций. C. J. Watson et al. [8] прогнозируют, что при пределе обнаружения ЧВА 0,01 % < 15 % люди в возрасте 80 лет и старше будут иметь клоны с двумя или более мутациями в одной клетке. Будущие исследования с использованием секвенирования нового поколения отдельных клеток могут ответить на этот вопрос.
МДС и клональная цитопения неопределенного значения или апластическая анемия (АА) имеют общий набор генетических изменений, часто приводящих к ОМЛ (табл. 3) [74].
Таблица 3. Сравнение характеристик различных гематологических заболеваний при КК
Table 3. Comparison of the characteristics of various hematological diseases with CH

Хотя мутации КК связаны с повышенным риском развития ОМЛ, выполнение скрининга периферической крови у всех людей пожилого возраста невыполнимо из-за сложности и высокой стоимости исследования. Тем не менее в результате расширения применения панелей секвенирования, КК уже определяется в различных клинических сценариях, в том числе в геномике опухолей. Данные исследований больных с гематологическими и солидными злокачественными новообразованиями и скриннинговых полногеномных анализов продемонстрировали наличие специфических мутаций в клонах кроветворных клеток [75],[76],[77]. Мутации, характерные для КК, также часто обнаруживают во время диагностики цитопений неясной этиологии. В настоящее время гены, мутации в которых встречаются при КК и связаны с риском трансформации в ОМЛ, включают DNMT3A, TET2, IDH1, IDH2, TP53, SRSF2, U2AF1, SF3B1, ZRSR2 и PPM1D [48],[49],[78],[79],[80]. Если у человека выявлены мутации в одном или нескольких из этих генов, целесообразно динамическое наблюдение за ним, мониторинг общего анализа крови. Не разработаны однозначные рекомендации относительно ЧВА, но при выборе лиц для мониторинга можно выделить следующие группы:
• мутации КК в IDH, TP53, генах сплайсосом при любом ЧВА;
• мутации КК в DNMT3A и TET2 с высоким ЧВА (> 10 %);
• мутации КК с клональной сложностью, включающей множественные мутации в генах.
Оптимальные сроки для последующего клинического наблюдения также неизвестны, но предлагается проводить мониторинг общего анализа крови с подсчетом лейкоцитарной формулы не реже двух раз в год. Рассматривается вариант более пристального наблюдения за людьми с мутациями DNMT3A и TET2 с ЧВА более 30 % и/или лицами с клональной сложностью, т. е. при обнаружении более 4 мутаций КК [14]. Большинство авторов процитированных работ считает, что клиническое применение их результатов состоит в том, что при выявлении мутаций КК, связанных с повышенным риском развития ОМЛ, следует начать мониторинг и периодические лабораторные исследования у этих людей.
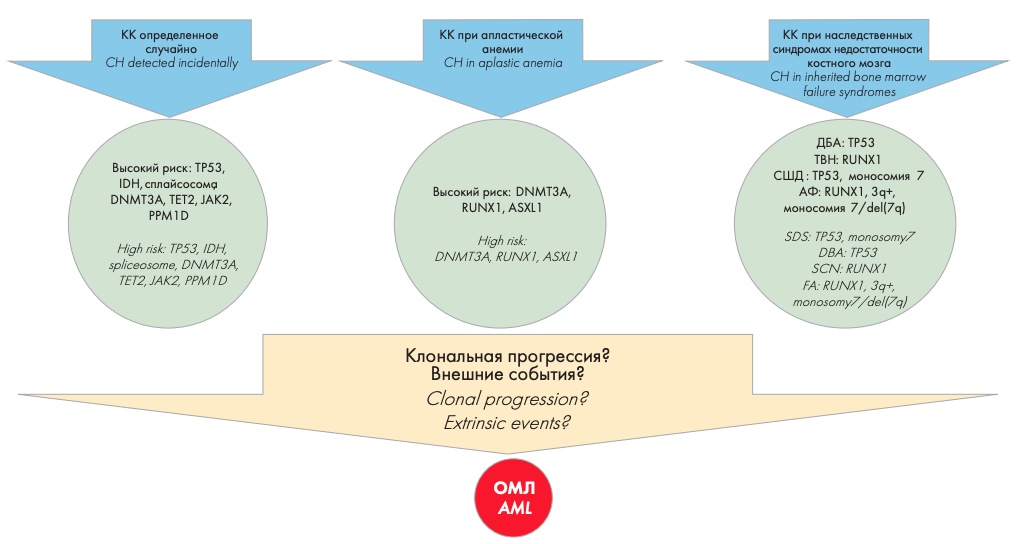
Рисунок 1. Связь мутаций КК высокого риска и риска развития ОМЛ. Мутации КК могут быть обнаружены случайно у больных во время диагностического обследования при цитопении или других заболеваниях. Тестирование на КК может проводиться при выявлении врожденных и приобретенных синдромов недостаточности костного мозга: синдром Швахмана — Даймонда (СШД), Даймонда — Блекфена анемия (ДБА), тяжелая врожденная нейтропения (ТВН); анемия Фанкони (АФ) (адаптировано из P. Desai и соавт. [14])
Figure 1. Association of high risk CH mutations and risk of AML development. CH mutations can be detected incidentally in patients during diagnostics of cytopenias or other pathologies. CH testing can also be done during work up of congenital and acquired bone marrow failure syndromes: Schwachman — Diamond Syndrome (SDS), Diamond — Blackfan Anemia (DBA), Severe Congenital Neutropenia (SCN), Fanconi Anemia (FA) (adapted from P. Desai et al. [14])
У людей с мутациями в генах IDH и TP53 риск развития ОМЛ не зависит от исходного размера клона [47]. Исходный уровень ЧВА ≥ 10 % связан с более высокой вероятностью развития ОМЛ, по сравнению с ЧВА < 10 % для мутаций в гене DNMT3A (ОШ — 4,8 против 2,5, p = 0,002), TET2 (ОШ — 20,4 против 3,6, p = 0,004) и сплайсосомных генах (ОШ — 14,1 против 3,4, p = 0,019). При серийном наблюдении за большинством клонов, ассоциированных с развитием ОМЛ, ЧВА увеличивалась еще до момента постановки диагноза заболевания. По данным S. Abelson et al. [48], риск развития ОМЛ увеличивался в 2 раза с увеличением ЧВА большинства генов на каждые 5 %, за исключением мутаций в генах DNMT3A и TET2, где пропорциональное увеличение риска было ниже. Размеры клонов более 30 % для DNMT3A и 10 % для TET2 были обнаружены только у 1 % лиц из контрольной группы. Однако некоторые исследователи не выявили различий в ЧВА в периферической крови людей, у которых в последствии развился ОМЛ, и теми, у которых этого не произошло [49]. Возможным объяснением этого феномена является меньший размер выборки и разницу во времени сбора крови до начала исследования, а также в момент установления диагноза ОМЛ.
S. Boettcher и соавт. [81] тестировали КК в условиях, когда СКК одного и того же человека подвергались разной степени воздействия: пролиферативному стрессу и внешнему влиянию, т. е. у лиц через длительное время после трансплантации аллогенных гемопоэтических стволовых клеток (алло-ТГСК) и их соответствующих родственных доноров (42 пары «донор — реципиент»). При среднем времени наблюдения с момента аллоТГСК в течение 16 лет (диапазон — 10–32 года) обнаружили в общей сложности 35 мутаций у 23 из 84 (27,4 %) участников исследования. КК выявили у 10 из 42 доноров (23,8 %) и 13 из 42 реципиентов (31 %). КК ассоциировалось с более старшим возрастом донора и реципиента. Идентифицировали 5 случаев трансплантации КК от донора, и в 1 случае КК трансформировалось в МДС как у донора, так и у реципиента. В 4 из 5 наблюдений отмечено увеличение размера клона у реципиентов по сравнению с донорами. Авторы охарактеризовали систему кроветворения у лиц с КК следующим образом: 1) КК постоянно присутствовали в миелоидных клетках, но различались по пенетрантности в В- и Т-клетках; 2) колониеобразующие единицы выявили клональную эволюцию или множественные независимые клоны у людей с множественными мутациями в КК; 3) укорочение теломер, определенное в гранулоцитах, предполагает примерно 20-летнее увеличение пролиферативного анамнеза СКК у реципиентов по сравнению с их донорами. Это исследование дает представление о долгосрочном поведении одних и тех же СКК человека и соответствующем развитии КК при различных условиях пролиферации.
В экспериментальной модели на мышах с Tet2-дефицитными макрофагами было показано, что атеросклероз и ишемическая болезнь сердца вызывали КК через изменение функции инфламмасом, приводившее к увеличению содержания провоспалительных цитокинов [82]. Обнаружена корреляция между мутациями гена DNMT3A и хронической болезнью «трансплантат против хозяина» после алло-ТГСК, что свидетельствует о роли КК в хронических воспалительных реакциях [83].
Предположено, что увеличение пролиферации СКК при воспалительных процессах у реципиентов может способствовать появлению клонального отбора и/или эволюции клонов КК [84]. При трансплантации аутологичных [85] или аллогенных СКК существующие донорские клоны КК могут приживаться и эволюционировать у реципиента, иногда приводя к опухолевой трансформации клеток донора [86].
Таким образом, впервые КК было выявлено у женщин, на основе принципа случайной инактивации Х-хромосомы почти 70 лет назад. За последнее время стало возможным проанализировать КК у 50 000 людейразноговозрастаиотследитьихдальнейшуюсудьбу, удалось установить размер клонов и рассчитать вероятность развития гематологических заболеваний, исходя из количества мутаций в определенных генах. Большинство генов, ассоциированных с КК, относится к внутренней регуляции СКК. Наличие КК связано с повышенным риском развития как первичного, так и вторичного, обусловленного предшествующей химиотерапией, ОМЛ. Мутации в генах TP53, IDH1/2, DNMT3A, TET2 и сплайсосомные мутации повышают риск развития de novo ОМЛ в 10 раз, тогда как мутации в генах PPM1D и TP53 при КК значительно повышают риск развития вторичного ОМЛ. Выявление больных с КК и наблюдение за ними позволят понять патогенез заболевания и разработать методы для предотвращения его развития. КК связано не только с риском развития заболеваний кроветворной системы, но и сердечно-сосудистых и других заболеваний.
1. Doulatov S., Notta F., Laurenti E., et al. Hematopoiesis: A human perspective. Cell Stem Cell. 2012; 10(2): 120–36. DOI: 10.1016/j.stem.2012.01.006.
2. Bonnet D. Biology of human bone marrow stem cells. Clin Exp Med. 2003; 3(3): 140–9. DOI: 10.1007/s10238-003-0017-9.
3. Phillips R.A. Hematopoietic stem cells: Concepts, assays, and controversies. Semin Immunol. 1991; 3(6): 337–47.
4. Siminovitch L., Mcculloch E.A., Till J.E. The distribution of colony-forming cells among spleen colonies. J Cell Physiol. 1963; 62: 327–36. DOI: 10.1002/jcp.1030620313.
5. Drize N.J., Keller J.R., Chertkov J.L. Local clonal analysis of the hematopoietic system shows that multiple small short-living clones maintain life-long hematopoiesis in reconstituted mice. Blood. 1996; 88(8): 2927–38.
6. Goyal S., Zandstra P.W. Stem cells: Chasing blood. Nature. 2015; 518(7540): 488–90. DOI: 10.1038/nature14203.
7. Carrelha J., Meng Y., Kettyle L.M., et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature. 2018; 554(7690): 106–11. DOI: 10.1038/nature25455.
8. Watson C.J., Papula A.L., Poon G.Y.P., et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science. 2020; 367(6485): 1449–54. DOI: 10.1126/science.aay9333.
9. Jones R.J., Armstrong S.A. Cancer stem cells in hematopoietic malignancies. Biol Blood Marrow Transplant. 2008; 14(1 Suppl 1): 12–6. DOI: 10.1016/j.bbmt.2007.10.012.
10. Shlush L.I. Age-related clonal hematopoiesis. Blood. 2018; 131(5): 496–504. DOI: 10.1182/blood-2017-07-746453.
11. Jaiswal S., Fontanillas P., Flannick J., et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014; 371(26): 2488–98. DOI: 10.1056/NEJMoa1408617.
12. Jaiswal S., Natarajan P., Silver A.J., et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017; 377(2): 111–21. DOI: 10.1056/NEJMoa1701719.
13. Genovese G., Kähler A.K., Handsaker R.E., et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014; 371(26): 2477–87. DOI: 10.1056/NEJMoa1409405.
14. Desai P., Hassane D., Roboz G.J. Clonal hematopoiesis and risk of acute myeloid leukemia. Best Pract Res Clin Haematol. 2019; 32(2): 177–85. DOI: 10.1016/j.beha.2019.05.007.
15. Adelman E.R., Figueroa M.E. Human hematopoiesis: aging and leukemogenic risk. Curr Opin Hematol. 2021; 28(1): 57–63. DOI: 10.1097/MOH.0000000000000622.
16. Ayachi S., Buscarlet M., Busque L. 60 years of clonal hematopoiesis research: From X-chromosome inactivation studies to the identification of driver mutations. Exp Hematol. 2020; 83: 2–11. DOI: 10.1016/j.exphem.2020.01.008.
17. Zink F., Stacey S.N., Norddahl G.L., et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017; 130(6): 742–52. DOI: 10.1182/blood-2017-02-769869.
18. Lyon M.F. Sex chromatin and gene action in the mammalian X-chromosome. Am J Hum Genet. 1962; 14(2): 135–48.
19. Fialkow P.J. The origin and development of human tumors studied with cell markers. N Engl J Med. 1974; 291(1): 26–35. DOI: 10.1056/NEJM197407042910109.
20. Fey M.F., Peter H.J., Hinds H.L., et al. Clonal analysis of human tumors with M27 beta, a highly informative polymorphic X chromosomal probe. J Clin Invest. 1992; 89(5): 1438–44. DOI: 10.1172/JCI115733.
21. Abkowitz J.L., Taboada M., Shelton G.H., et al. An X chromosome gene regulates hematopoietic stem cell kinetics. Proc Natl Acad Sci U S A. 1998; 95(7): 3862–6. DOI: 10.1073/pnas.95.7.3862.
22. Busque L., Patel J.P., Figueroa M.E., et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012; 44(11): 1179–81. DOI: 10.1038/ng.2413.
23. Laurie C.C., Laurie C.A., Rice K., et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012; 44(6): 642–50. DOI: 10.1038/ng.2271.
24. Xie M., Lu C., Wang J., et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014; 20(12): 1472–8. DOI: 10.1038/nm.3733.
25. Steensma D.P., Ebert B.L. Clonal hematopoiesis as a model for premalignant changes during aging. Exp Hematol. 2020; 83: 48–56. DOI: 10.1016/j.exphem.2019.12.001.
26. Luis T.C., Wilkinson A.C., Beerman I., et al. Biological implications of clonal hematopoiesis. Exp Hematol. 2019; 77: 1–5. DOI: 10.1016/j.exphem.2019.08.004.
27. Zink F., Stacey S.N., Norddahl G.L., et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017; 130(6): 742–52. DOI: 10.1182/blood-2017-02-769869.
28. Buscarlet M., Provost S., Zada Y.F., et al. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood. 2018; 132(3): 277–80. DOI: 10.1182/blood-2018-01-829937.
29. de Haan G., Lazare S.S. Aging of hematopoietic stem cells. Blood. 2018; 131(5): 479–87. DOI: 10.1182/blood-2017-06-746412.
30. Chambers S.M., Goodell M.A. Hematopoietic stem cell aging: Wrinkles in stem cell potential. Stem Cell Rev. 2007; 3(3): 201–11. DOI: 10.1007/s12015-007-0027-1.
31. Snoeck H.-W. Aging of the hematopoietic system. Curr Opin Hematol. 2013; 20(4): 355–61. DOI: 10.1097/MOH.0b013e3283623c77.
32. Beerman I. Accumulation of DNA damage in the aged hematopoietic stem cell compartment. Semin Hematol. 2017; 54(1): 12–8. DOI: 10.1053/j.seminhematol.2016.11.001.
33. Mollica L., Fleury I., Belisle C., et al. No association between telomere length and blood cell counts in elderly individuals. J Gerontol A Biol Sci Med Sci. 2009; 64(9): 965–7. DOI: 10.1093/gerona/glp065.
34. Ho T.T., Warr M.R., Adelman E.R., et al. Autophagy maintains the metabolism and function of young and old stem cells. Nature. 2017; 543(7644): 205–10. DOI: 10.1038/nature21388.
35. Kramer A., Challen G.A. The epigenetic basis of hematopoietic stem cell aging. Semin Hematol. 2017; 54(1): 19–24. DOI: 10.1053/j.seminhematol.2016.10.006.
36. Osorio F.G., Rosendahl Huber A., Oka R., et al. Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Rep. 2018; 25(9): 2308–16.e4. DOI: 10.1016/j.celrep.2018.11.014.
37. Tsai F.D., Lindsley R.C. Clonal hematopoiesis in the inherited bone marrow failure syndromes. Blood. 2020; 136(14): 1615–22. DOI: 10.1182/blood.2019000990.
38. Holstege H., Pfeiffer W., Sie D., et al. Somatic mutations found in the healthy blood compartment of a 115-yr-old woman demonstrate oligoclonal hematopoiesis. Genome Res. 2014; 24(5): 733–42. DOI: 10.1101/gr.162131.113.
39. Fabre M.A., McKerrell T., Zwiebel M., et al. Concordance for clonal hematopoiesis is limited in elderly twins. Blood. 2020; 135(4): 269–73. DOI: 10.1182/blood.2019001807.
40. Fabre M.A., Vassiliou G.S. Home and away: Clonal hematopoiesis in sibling transplants. Blood. 2020; 135(18): 1511–2. DOI: 10.1182/blood.2020005717.
41. Hansen J.W., Pedersen D.A., Larsen L.A., et al. Clonal hematopoiesis in elderly twins: Concordance, discordance, and mortality. Blood. 2020; 135(4): 261–8. DOI: 10.1182/blood.2019001793.
42. Shlush L.I. Clonal hematopoiesis sees Twin Peaks. Blood. 2020; 135(4): 235–6. DOI: 10.1182/blood.2019003869.
43. Valent P., Kern W., Hoermann G., et al. Clonal Hematopoiesis with Oncogenic Potential (CHOP): Separation from CHIP and roads to AML. Int J Mol Sci. 2019; 20(3): 789. DOI: 10.3390/ijms20030789.
44. Warren J.T., Link D.C. Clonal hematopoiesis and risk for hematologic malignancy. Blood. 2020; 136(14): 1599–605. DOI: 10.1182/blood.2019000991.
45. Loh P.-R., Genovese G., Handsaker R.E., et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature. 2018; 559(7714): 350–5. DOI: 10.1038/s41586-018-0321-x.
46. Döhner H., Weisdorf D.J., Bloomfield C.D. Acute myeloid leukemia. N Engl J Med. 2015; 373(12): 1136–52. DOI: 10.1056/NEJMra1406184.
47. Desai P., Mencia-Trinchant N., Savenkov O., et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. 2018; 24(7): 1015–23. DOI: 10.1038/s41591-018-0081-z.
48. Abelson S., Collord G., Ng S.W.K., et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018; 559(7714): 400–4. DOI: 10.1038/s41586-018-0317-6.
49. Young A.L., Tong R.S., Birmann B.M., et al. Clonal hematopoiesis and risk of acute myeloid leukemia. Haematologica. 2019; 104(12): 2410–7. DOI: 10.3324/haematol.2018.215269.
50. Steensma D.P., Bejar R., Jaiswal S., et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015; 126(1): 9–16. DOI: 10.1182/blood-2015-03-631747
51. Young A.L., Challen G.A., Birmann B.M., et al. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016; 7: 12484. DOI: 10.1038/ncomms12484.
52. Higgins A., Shah M.V. Genetic and genomic landscape of secondary and therapy-related acute myeloid leukemia. Genes (Basel). 2020; 11(7): 749. DOI: 10.3390/genes11070749.
53. Patel J.P., Gönen M., Figueroa M.E., et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012; 366(12): 1079–89. DOI: 10.1056/NEJMoa1112304.
54. Buscarlet M., Provost S., Zada Y.F., et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. 2017; 130(6): 753–62. DOI: 10.1182/blood-2017-04-777029.
55. Genovese G., Kähler A.K., Handsaker R.E., et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014; 371(26): 2477–87. DOI: 10.1056/NEJMoa1409405.
56. Jaiswal S., Fontanillas P., Flannick J., et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014; 371(26): 2488–98. DOI: 10.1056/NEJMoa1408617.
57. Lindsley R.C., Mar B.G., Mazzola E., et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015; 125(9): 1367–76. DOI: 10.1182/blood-2014-11-610543.
58. Heuser M., Thol F., Ganser A. Clonal hematopoiesis of indeterminate potential. Dtsch Arztebl Int. 2016; 113(18): 317–22. DOI: 10.3238/arztebl.2016.0317.
59. Kadia T.M., Jain P., Ravandi F., et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer. 2016; 122(22): 3484–91. DOI: 10.1002/cncr.30203.
60. Metzeler K.H., Herold T., Rothenberg-Thurley M., et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016; 128(5): 686–98. DOI: 10.1182/blood-2016-01-693879.
61. Mason C.C., Khorashad J.S., Tantravahi S.K., et al. Age-related mutations and chronic myelomonocytic leukemia. Leukemia. 2016; 30(4): 906–13. DOI: 10.1038/leu.2015.337.
62. Sperling A.S., Gibson C.J., Ebert B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017; 17(1): 5–19. DOI: 10.1038/nrc.2016.112.
63. Takahashi S. Epigenetic aberrations in myeloid malignancies (Review). Int J Mol Med. 2013; 32(3): 532–8. DOI: 10.3892/ijmm.2013.1417.
64. Metzeler K.H., Maharry K., Radmacher M.D., et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: A Cancer and Leukemia Group B study. J Clin Oncol. 2011; 29(10): 1373–81. DOI: 10.1200/JCO.2010.32.7742.
65. Figueroa M.E., Lugthart S., Li Y., et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010; 17(1): 13–27. DOI: 10.1016/j.ccr.2009.11.020.
66. Cai Z., Kotzin J.J., Ramdas B., et al. Inhibition of infl ammatory signaling in Tet2 mutant preleukemic cells mitigates stress-induced abnormalities and clonal hematopoiesis. Cell Stem Cell. 2018; 23(6): 833–49.e5. DOI: 10.1016/j.stem.2018.10.013.
67. Katoh M. Functional and cancer genomics of ASXL family members. Br J Cancer. 2013; 109(2): 299–306. DOI: 10.1038/bjc.2013.281.
68. Papaemmanuil E., Gerstung M., Malcovati L., et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013; 122(22): 3616–27; quiz 3699. DOI: 10.1182/blood-2013-08-518886.
69. Itzykson R., Kosmider O., Renneville A., et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013; 31(19): 2428–36. DOI: 10.1200/JCO.2012.47.3314.
70. Papaemmanuil E., Gerstung M., Bullinger L., et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016; 374(23): 2209–21. DOI: 10.1056/NEJMoa1516192.
71. Volkert S., Kohlmann A., Schnittger S., et al. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes Chromosomes Cancer. 2014; 53(5): 402–10. DOI: 10.1002/gcc.22151.
72. Sebaa A., Ades L., Baran-Marzack F., et al. Incidence of 17p deletions and TP53 mutation in myelodysplastic syndrome and acute myeloid leukemia with 5q deletion. Genes Chromosomes Cancer. 2012; 51(12): 1086–92. DOI: 10.1002/gcc.21993.
73. Jones A.V., Kreil S., Zoi K., et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005; 106(6): 2162–8. DOI: 10.1182/blood-2005-03-1320.
74. Hartmann L., Metzeler K.H. Clonal hematopoiesis and preleukemia – Genetics, biology, and clinical implications. Genes Chromosomes Cancer. 2019; 58(12): 828–38. DOI: 10.1002/gcc.22756.
75. Severson E.A., Riedlinger G.M., Connelly C.F., et al. Detection of clonal hematopoiesis of indeterminate potential in clinical sequencing of solid tumor specimens. Blood. 2018; 131(22): 2501–5. DOI: 10.1182/blood-2018-03-840629.
76. Ptashkin R.N., Mandelker D.L., Coombs C.C., et al. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA Oncol. 2018; 4(11): 1589–93. DOI: 10.1001/jamaoncol.2018.2297.
77. Phallen J., Sausen M., Adleff V., et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med. 2017; 9(403): eaan2445. DOI: 10.1126/scitranslmed.aan2415.
78. Gillis N.K., Ball M., Zhang Q., et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: A proof-of-concept, case-control study. Lancet Oncol. 2017; 18(1): 112–21. DOI: 10.1016/S1470-2045(16)30627-1.
79. Gibson C.J., Lindsley R.C., Tchekmedyian V., et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017; 35(14): 1598–605. DOI: 10.1200/JCO.2016.71.6712.
80. Mouhieddine T.H., Sperling A.S., Redd R., et al. Clonal hematopoiesis is associated with adverse outcomes in multiple myeloma patients undergoing transplant. Nat Commun. 2020; 11(1): 2996. DOI: 10.1038/s41467-020-16805-5.
81. Boettcher S., Wilk C.M., Singer J., et al. Clonal hematopoiesis in donors and long-term survivors of related allogeneic hematopoietic stem cell transplantation. Blood. 2020; 135(18): 1548–59. DOI: 10.1182/blood.2019003079.
82. Arends C.M., Weiss M., Christen F., et al. Clonal hematopoiesis in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Haematologica. 2020; 105(6): e264–7. DOI: 10.3324/haematol.2019.223305.
83. Frick M., Chan W., Arends C.M., et al. Role of donor clonal hematopoiesis in allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. 2019; 37(5): 375–85. DOI: 10.1200/JCO.2018.79.2184.
84. Takizawa H., Boettcher S., Manz M.G. Demand-adapted regulation of early hematopoiesis in infection and infl ammation. Blood. 2012; 119(13): 2991–3002. DOI: 10.1182/blood-2011-12-380113.
85. Chitre S., Stölzel F., Cuthill K., et al. Clonal hematopoiesis in patients with multiple myeloma undergoing autologous stem cell transplantation. Leukemia. 2018; 32(9): 2020–4. DOI: 10.1038/s41375-018-0208-8.
86. Kato M., Yamashita T., Suzuki R., et al. Donor cell-derived hematological malignancy: A survey by the Japan Society for Hematopoietic Cell Transplantation. Leukemia. 2016; 30(8): 1742–5. DOI: 10.1038/leu.2016.23.
Петинати Наталия Арнольдовна, кандидат медицинских наук, старший научный сотрудник лаборатории физиологии кроветворения
125167, Москва
Дризе Нина Иосифовна, доктор биологических наук, профессор, заведующая лабораторией физиологии кроветворения
125167, Москва
Петинати Н.А., Дризе Н.И. Клональное кроветворение и его роль в развитии гематологических заболеваний. Гематология и трансфузиология. 2021;66(4):580-592. https://doi.org/10.35754/0234-5730-2021-66-4-580-592
Petinati N.A., Drize N.J. Clonal hematopoiesis and its role in the development of hematological diseases. Russian journal of hematology and transfusiology. 2021;66(4):580-592. (In Russ.) https://doi.org/10.35754/0234-5730-2021-66-4-580-592
![]()
125167, Москва, Новый Зыковский проезд, 4
ФГБУ «НМИЦ гематологии» Минздрава России
тел.: 8-926-816-3887
e-mail: o.levchenko@htjournal.ru






































