

Содержание
Перейти к:
 Е. А. Михайлова,
З. Т. Фидарова,
В. В. Троицкая,
Г. А. Клясова,
А. Д. Кулагин,
Е. В. Воронова,
В. Н. Двирнык,
И. В. Гальцева,
А. М. Ковригина,
Т. Н. Обухова,
Т. В. Гапонова,
Е. Н. Паровичникова,
В. Г. Савченко
Е. А. Михайлова,
З. Т. Фидарова,
В. В. Троицкая,
Г. А. Клясова,
А. Д. Кулагин,
Е. В. Воронова,
В. Н. Двирнык,
И. В. Гальцева,
А. М. Ковригина,
Т. Н. Обухова,
Т. В. Гапонова,
Е. Н. Паровичникова,
В. Г. Савченко https://doi.org/10.35754/0234-5730-2020-65-2-208-226
Перейти к:
Введение: по инициативе российского Национального гематологического общества исследовательской группой по изучению идиопатической апластической анемии разработаны клинические рекомендации по диагностике и лечению идиопатической апластической анемии.
Целью настоящих рекомендаций является стандартизация диагностических и лечебных подходов к лечению приобретенной апластической анемии в России.
Методы. Использовавшиеся методологические подходы основаны на принципах доказательной медицины, в их основе лежат рекомендации Российского совета экспертов по диагностике и лечению больных идиопатической апластической анемией, российский и международный опыт ведения больных, рекомендации европейской группы по изучению апластической анемии.
Результат. Представлен новый доработанный и дополненный вариант национальных клинических рекомендаций.
Заключение. Рекомендации предназначены для врачей различных специальностей, администраторов здравоохранения, студентов медицинских учебных заведений.
Конфликт интересов: авторы заявляют об отсутствии конфликта интересов.
Финансирование: работа не имела спонсорской поддержки.
Михайлова Е.А., Фидарова З.Т., Троицкая В.В., Клясова Г.А., Кулагин А.Д., Воронова Е.В., Двирнык В.Н., Гальцева И.В., Ковригина А.М., Обухова Т.Н., Гапонова Т.В., Паровичникова Е.Н., Савченко В.Г. Клинические рекомендации по диагностике и лечению апластической анемии (редакция 2019 г.). Гематология и трансфузиология. 2020;65(2):208-226. https://doi.org/10.35754/0234-5730-2020-65-2-208-226
Mihailova E.A., Fidarova Z.T., Troitskaya V.V., Klyasova G.A., Kulagin A.D., Voronova E.V., Dvirnyk V.N., Galtseva I.V., Kovrigina A.M., Obukhova T.N., Gapanova T.V., Parovichnikova E.N., Savchenko V.G. Clinical recommendations for the diagnosis and treatment of aplastic anemia (2019 edition). Russian journal of hematology and transfusiology. 2020;65(2):208-226. (In Russ.) https://doi.org/10.35754/0234-5730-2020-65-2-208-226
Агонисты тромбопоэтиновых рецепторов стимулируют гемопоэз, индуцируют пролиферацию и диффе- ренцировку гемопоэтических клеток-предшественниц.
Клинико-гематологическое улучшение — улучшение показателей гемограммы (гемоглобин >80,0 г/л, гранулоциты >0,5 х 10 9/л для тяжелой формы АА (ТАА) или гранулоциты >1,0 х 10 9/л для нетяжелой АА (НАА), тромбоциты >20,0 х 10 9/л и исчезновение или значительное уменьшение зависимости от трансфузий компонентов крови.
Клон пароксизмальной ночной гемоглобинурии (ПНГ-клон) — клон стволовой клетки крови, развившийся в результате мутации в PIG-A гене, приводящей к нарушению синтеза гликозилфосфатидилинозито- ла — якоря, с помощью которого к мембранам клеток крепятся белки, защищающие их от воздействия компонентов системы комплемента.
Комбинированная иммуносупрессивная терапия (ИСТ) больных АА проводится по протоколу, включающему лошадиный антитимоцитарный глобулин и циклоспорин.
Программное лечение больных апластической анемией — комплекс лечебных мероприятий, проводимых поэтапно, начиная с момента диагностики заболевания, осуществляемый в определенном алгоритме, включающий антитимоцитарный глобулин, циклоспорин А, при необходимости — элтромбопаг, повторные курсы антитимоцитарного глобулина (АТГ) и другие методы терапии, позволяющие добиться длительной выживаемости больных.
Ремиссия (полная или частичная) — полная или частичная нормализация показателей гемограммы (гемоглобин >100 г/л, гранулоциты >1,5 х 109/л, тромбоциты >100 х 109/л) и отсутствие потребности в заместительной терапии компонентами крови.
Рефрактерная апластическая анемия диагностируется в случае отсутствия эффекта от проводимой комбинированной ИСТ через 6 месяцев от начала лечения или после второго курса АТГ.
Хелаторная терапия назначается больным с диагностированной перегрузкой железом вследствие многочисленных гемотрансфузий.
Апластическая анемия (АА) — заболевание системы крови, характеризующееся панцитопенией, обусловленной аплазией костного мозга, связанной с нарушением иммунных механизмов регуляции кроветворения, количественным дефицитом и функциональными дефектами стволовых кроветворных клеток.
Одним из ведущих механизмов поражения кроветворения при АА считается иммунная агрессия, направленная на клетки-предшественницы гемопоэза [1]. Костномозговая недостаточность при АА развивается в результате подавления пролиферации гемо- поэтических клеток-предшественниц активированными Т-лимфоцитами и естественными киллерами. Активация Т-лимфоцитов, экспансия цитотоксических Т-клонов и выброс медиаторов иммунной супрессии кроветворения (интерферон γ, фактор некроза опухолей α и другие цитокины) или стимулирующих пролиферацию и активацию Т-лимфоцитов (интерлейкин-2) приводят к нарушению процессов пролиферации и к стимуляции апоптоза клеток-предшественниц. Происходит значительное уменьшение пула гемопоэтических клеток и развитие аплазии костного мозга [2]. Определенную роль в патогенезе АА могут играть и другие механизмы развития костномозговой недостаточности, связанные с нарушениями микроокружения (стромы костного мозга) [3] и с клональными перестройками в стволовых клетках крови (СКК) в результате хромосомных аномалий, геномной нестабильности, истощения теломерных участков ДНК в СКК и соматическими мутациями, характерными для миелоидных заболеваний [4—6].
Уменьшение пула гемопоэтических клеток костного мозга сопровождается нарушением обмена железа и отложением токсического железа в костном мозге, миокарде, печени, эндокринных и половых органах, что вызывает нарушение функции этих органов. Нарушение обмена железа, гемосидероз внутренних органов усугубляются длительной гемотрансфузионной терапией, необходимой у большинства больных АА [4].
Кроме того, течение АА может осложниться развитием таких клональных заболеваний, как пароксизмальная ночная гемоглобинурия (ПНГ), миелодиспластический синдром (МДС), острый миелоидный лейкоз (ОМЛ). Частота развития клональных осложнений может достигать 32% в течение 10 лет [3, 4]. Появление клонального кроветворения может быть выявлено и на более ранних этапах течения АА. В первую очередь речь идет об АА, протекающей с ПНГ- клоном [7, 8]. При этом выявление клона с дефицитом гликозилфосфатидилинозитол-заякоренных белков (ГФИ-белков) не означает развитие ПНГ как самостоятельного заболевания с картиной классического внутрисосудистого гемолиза. Размер ПНГ-клона в процессе течения АА может меняться: увеличиваться вплоть до трансформации в классическую ПНГ или уменьшаться до полного исчезновения. Эволюция в классическую ПНГ, по данным различных авторов, составляет 11—17% [9—12].
АА, по данным эпидемиологических исследований, встречается с различной частотой в таких регионах, как Европа, Северная Америка, Дальний и Ближний Восток, при этом в европейских странах заболеваемость АА составляет 2 случая на 1 млн населения в год, этот показатель колеблется в зависимости от конкретной страны от 0,6 до 3 и более [13].
Другие апластические анемии (D61)
D61.2 | АА, вызванная другими внешними агентами |
D61.3 | Идиопатическая АА |
D61.8 | Другие уточненные АА |
D61.9 | АА неуточненная |
Приобретенная АА |
|
|---|---|
1. Идиопатическая АА | Этиологический фактор не идентифицирован |
2. Вирус-ассоциированные | Вирусные гепатиты, вирус Эпштейна — Барр, вирус иммунодефицита человека, парвовирус В19 у больных с иммунодефицитом |
3. Лекарственные и токсические | Хлорамфеникол, бензин, радиация |
4. Вторичная АА (апластический синдром) | На фоне иммунных заболеваний: • системная красная волчанка и другие коллагенозы • гипогаммаглобулинемия (общая вариабельная иммунная недостаточность — ОВИН), синдром Ниймеген, Х-сцепленный лимфопролиферативный синдром) |
Тимома и карцинома тимуса | |
ПНГ | |
Злокачественные лимфопролиферативные заболевания (Т-лейкоз из больших гранулированных лимфоцитов, лимфома с поражением селезенки) | |
Миелодиспластический синдром |
Диагноз АА устанавливается на основании клинических проявлений болезни и данных лабораторного обследования (Приложение № 1). Основными клиническими проявлениями болезни являются анемический, геморрагический синдромы, а также тяжелые инфекционные осложнения.
Критерии диагноза:
Критерии тяжести АА:
При определении тяжести АА учитываются результаты не менее трех анализов периферической крови на момент диагностики заболевания до начала лечения [14, 15].
Рекомендуется выделять варианты течения АА:
Жалобы анемического характера, развитие геморрагического синдрома различной интенсивности и инфекционных осложнений на фоне глубокой трехростковой цитопении — определяют дебют клинических проявлений АА. Данные проявления могут развиться остро или постепенно нарастать, в зависимости от тяжести заболевания.
Рекомендуется при АА из анамнестических данных выявлять связь с возможными токсическими, лекарственными агентами или ассоциацию с вирусными гепатитами В и С. Необходим тщательный сбор семейного анамнеза для исключения врожденных аномалий, а также уточнять наличие сиблингов (родных братьев и/или сестер) для рассмотрения возможности проведения трансплантации гемопоэтических стволовых клеток (ТГСК). В анамнезе заболевания должны быть описаны все эпизоды инфекционных осложнений, проведенная антибиотическая терапия, данные бактериологических посевов в течение последних 2—3 лет. Необходимо полное описание частоты и потребности в проводимой гемотрансфузионной терапии и возможных осложнений на ее фоне [15].
При АА осмотр включает измерение роста и массы тела, температуры тела; оценку состояния костно-суставной системы; выявление признаков геморрагического синдрома; отсутствие гепатоспленомегалии, лим- фоаденопатии; наличие признаков дисфункции сердца, легких, печени, почек, органов эндокринной системы.
Рекомендуется при физикальном обследовании обращать внимание на аномалии, характерные для конституциональных форм АА (пигментация кожных покровов, дистрофия ногтей, лейкоплакия слизистых, аномалии развития глаз, аномалии зубов, раннее поседение и выпадение волос, гипогонадизм и т. д.) [15—17].
Рекомендуется выполнение следующих диагностических исследований (Приложение № 6).
Рекомендуется выполнение следующих инструментальных диагностических методов [14].
Контрольные обследования больного (общий анализ периферической крови, биохимическое исследование крови, общий анализ мочи) проводятся 1 раз в неделю до достижения ответа, в дальнейшем — 1 раз в месяц. Стернальную пункцию и трепанобиопсию выполняют каждые 6—12 месяцев; иммунофенотипирование клеток периферической крови с целью определения ПНГ-клона — каждые 6—12 месяцев [15, 20, 21].
Программа лечения взрослых больных АА — это комбинированная ИСТ, проводимая с использованием двух основных препаратов, обладающих выраженным иммуносупрессивным действием: АТГ и циклоспорина А, и/или ТГСК [33—36]. Современная патогенетическая терапия больных АА может включать кроме препаратов с иммуносупрессивным действием (АТГ, циклоспорин А) лекарственные препараты, направленные на активацию пролиферации клеток-предшественниц кроветворения и одновременное подавление активации цитотоксических клеток. Многочисленными клиническими исследованиями было показано значительное улучшение гематологических показателей под влиянием элтромбопага [37]. Элтромбопаг является агонистом тромбопоэтиновых рецепторов, которые локализуются не только на мегакариоцитах, но и на стволовых клетках крови. Элтромбопаг связывается с трансмембранным доменом тромбопоэтиновых рецепторов, не конкурируя с эндогенным тромбопоэтином, и обладает иммуномодулирующими свойствами посредством активации Т-регуляторных клеток. Кроме того, элтромбопаг обладает хелаторной активностью и способствует выведению токсического железа из клеток печени, сердца и других внутренних органов при перегрузке железом у больных АА, зависимых от трансфузий донорских эритроцитов [37]. Клиническое использование элтромбопага может сопровождаться моно-, би-, трехлинейным гематологическим ответом у больных рефрактерной АА, а его использование в программах комбинированной терапии АА достоверно повышает частоту достижения полного ответа и общую выживаемость больных [37]. Программа лечения может включать и другие терапевтические воздействия, в частности хелаторную терапию, а также спленэктомию [5, 14, 38]. Огромную роль в реализации программы лечения больных АА играет заместительная гемотрансфузионная терапия: трансфузии донорских клеток крови (эритроцитная взвесь и концентрат тромбоцитов) [18, 39].
ТГСК в рамках алгоритма лечения больных АА занимает определенное место: наличие HLA-идентичного родственного донора, молодой возраст, короткий гемотрансфузионный анамнез — условия, при которых ТГСК может рассматриваться как терапия выбора (терапия 1-й линии) [40]. Существенным недостатком этого метода является ограниченная возможность применения, связанная с отсутствием родственного донора костного мозга у большинства взрослых больных. Тем не менее ТГСК в настоящее время рассматривается как терапия выбора на первом этапе лечения молодых больных тяжелой АА, имеющих HLA-идентичного родственного донора костного мозга [41, 42]. Поэтому уже при диагностировании АА необходимо выполнение HLA-типирования больного и поиск возможного родственного донора. Абсолютным показанием к проведению ТГСК при АА на любом этапе течения болезни является выявление при цитогенетическом исследовании неблагоприятных хромосомных аберраций и прежде всего моносомии 7- й хромосомы, свидетельствующих о прогрессии в МДС или ОМЛ [43, 44].
Совершенствование ИСТ, реализация программы на ранних этапах течения болезни позволили значительно повысить эффективность лечения АА: вероятность длительной выживаемости больных АА возросла до 80—90% [33]. Эффективность лечения зависит от своевременной диагностики АА, тяжести заболевания, возраста больного, сопутствующей патологии и возможности проведения комбинированной ИСТ или ТГСК уже на первых этапах лечения [14, 47, 48]. Использование при лечении больных АА глюкокор- тикоидных гормонов как основного метода терапии, неконтролируемой длительной монотерапии циклоспорином А (более 6 месяцев) и необоснованное применение колониестимулирующих факторов создают неблагоприятные условия для начала комбинированной ИСТ и ухудшают ее эффективность [14, 15].
Анализ эффективности комбинированной ИСТ взрослых больных АА, представленный в систематических обзорах клинических исследований в 2010—2019 гг., позволяет рекомендовать разработанный алгоритм лечения как основной протокол ИСТ взрослых больных АА [15, 45-48].
Оно начинается с момента диагностики заболевания и выбора терапевтической тактики (ТГСК или комбинированная ИСТ) (Приложение № 3).
Это комплекс лечебных мероприятий, проводимых поэтапно, включающий АТГ, циклоспорин А, при необходимости — повторные курсы АТГ, и другие методы ИСТ при рефрактерной АА, позволяющий добиться длительной выживаемости больных (Приложение № 4).
Терапия больных АА, протекающей с ПНГ-клоном и ПНГ-синдромом, определяется наличием аплазии костного мозга и выраженностью синдромов, характерных для АА, и проводится по протоколам лечения АА.
Противопоказаниями к проведению комбинированной ИСТ служат тяжелые соматические заболевания, сопровождающиеся сердечно-сосудистой, почечной, печеночной, дыхательной недостаточностью.
Геморрагический синдром и инфекционные осложнения следует рассматривать как временные противопоказания, которые должны быть купированы до начала ИСТ циклоспорином А или АТГ.
В случае тяжелых инфекционных осложнений (сепсис, пневмония) ИСТ должна предшествовать интенсивная противоинфекционная терапия, проводимая с учетом возбудителя (бактерии, грибы, вирусы). АТГ или циклоспорин назначаются через 5-7 дней после нормализации температуры и исчезновения клинической симптоматики.
При наличии геморрагического синдрома циклоспорин может быть назначен параллельно с заместительной терапией концентратом тромбоцитов.
Курс терапии циклоспорином у больных АА продолжают 24 месяца (не менее 12 месяцев после достижения ремиссии). Отмену препарата осуществляют медленно, по 50 мг в день каждые две недели.
Курс терапии АТГ соответствует протоколам № 1 (Приложение № 5). В течение первых 21-28 дней курса больной находится в асептических условиях одноместной палаты.
Перед началом курса устанавливают центральный венозный катетер.
Аллергические и анафилактические реакции во время введения АТГ — озноб, лихорадка, эритема- тозная или уртикарная сыпь — встречаются у 30-60% больных. В этих случаях увеличивают дозы вводимых глюкокортикоидных гормонов и антигистаминных препаратов в 1,5-2 раза и по возможности продолжают введение АТГ. Значительно реже (у 2-3% больных) развиваются бронхоспазм, отек Квинке, артериальная гипотензия. В этих случаях прекращают введение АТГ и проводят соответствующее лечение.
Сывороточная болезнь обычно развивается на 7-14-й день (5-20-й день) от начала терапии более чем у 50% больных. Лихорадка, папулезные высыпания на коже, кожный зуд, полиартралгии, миалгии, головная боль, тошнота — наиболее часто встречающиеся симптомы сывороточной болезни; реже наблюдаются транзитор- ное повышение активности аминотрансфераз в сыворотке, повышение артериального давления, желудочно-кишечные расстройства; в 1-2% случаев могут иметь место эпилептические судороги. Назначение антигистаминных препаратов и глюкокортикоидных гормонов (преднизолон 30-60 мг в сутки) в течение 1-2-х недель, как правило, купирует сывороточную болезнь. В тяжелых случаях проводят сеансы плазмафе- реза. Для предупреждения развития тяжелых аллергических осложнений рекомендуется непосредственно перед введением АТГ обязательно проводить внутрикожные тесты на чувствительность больного к препарату. Усиление геморрагического синдрома при введении или после введения АТГ требует интенсивной заместительной терапии трансфузиями концентрата тромбоцитов и свежезамороженной плазмы.
Наиболее часто встречающимся осложнением является нарушение функции почек, связанное с нефро- токсичностью препарата; при этом наблюдаются повышение содержания креатинина в сыворотке, периферические отеки и олигурия. Нередко повышается артериальное давление, появляются тремор пальцев рук, парестезии, головные боли, в редких случаях — энцефалопатия. У трети больных отмечается гиперплазия десен. Тошнота, рвота, боли в животе, диарея встречаются в 10-13% случаев. Гипербилирубинемия обнаруживается у 30% больных, но повышение сывороточной активности аминотрансфераз наблюдается значительно реже. Могут иметь место электролитные нарушения: гиперкалиемия, гипомагниемия. В некоторых случаях наблюдаются гинекомастия, гипертрихоз, а также аллергические реакции (аллергическая сыпь). Чаще всего перечисленные осложнения появляются в результате приема максимальной дозы циклоспорина (10 мг/кг в сутки) в первые несколько дней терапии. Токсические осложнения терапии циклоспорином корректируются уменьшением суточной дозы (на 25-50%) или временной отменой препарата. В ряде случаев необходима сопутствующая симптоматическая терапия.
Инфекционные осложнения при проведении ИСТ возникают у большинства больных АА. Как правило, вначале инфекционный процесс обусловлен бактериями, но в дальнейшем, по мере удлинения периода гранулоцитопении и усиления иммуносупрессии, доминируют оппортунистические инфекции, обусловленные грибами, прежде AspergiLLus spp., пневмоцистами, герпесвирусами [49]. К особенностям инфекционных осложнений, регистрируемых при АА, относят высокую частоту выявления возбудителя (микробиологически подтвержденные инфекции достигают 75%) и преобладание смешанной микрофлоры в этиологии инфекционного процесса. Инфекции, вызванные сочетанием микроорганизмов, могут быть как в первые дни инфекционного процесса, так и возникать при проводимой противомикробной терапии.
В первые сутки единственным симптомом инфекции бывает лишь повышение температуры, а информацию о наличии бактериемии или пневмонии у больных с нейтропенией (гранулоцитопенией) удается получить не раньше, чем на второй день инфекционного эпизода. Лихорадка при нейтропении расценивается как инфекционная, если температура тела поднимается выше 38 °С.
Проведение профилактики возможных осложнений начинают за 2 дня до начала терапии и продолжают 2 недели после окончания курса.
Проведение хелаторной терапии необходимо при концентрации ферритина сыворотки более 1000 нг/мл. При сочетанном применении с циклоспорином А необходим тщательный мониторинг состояния функции почек.
Спленэктомия лапароскопическим доступом может быть выполнена в случае необходимости проведения дифференциальной диагностики. Не рекомендуется проведение спленэктомии у больных с размером ПНГ- клона более 10% и/или наличием ПНГ-синдрома: клинические наблюдения свидетельствуют о быстрой прогрессии в гемолитическую форму ПНГ с характерными тяжелыми тромботическими осложнениями. Не рекомендуется проведение спленэктомии у больных с выявленными цитогенетическими аномалиями: высокая частота трансформации в МДС/ОМЛ.
Эффективность лечения оценивается по следующим критериям: клинико-гематологическое улучшение и отсутствие трансфузионной зависимости, частичная ремиссия и полная ремиссия.
В течение первого года контрольное обследование больных должно проводиться каждые 3 месяца, в дальнейшем — каждые 6 месяцев до достижения полного ответа на лечение. Далее — ежегодно. Отсутствие ответа на лечение (1-й курс АТГ + циклоспорин А) через 3-6 месяцев от начала лечения свидетельствует о рефрактерном течении болезни.
Таблица 1. Варианты ответа и динамика показателей гемограммы
Table 1. Response options and dynamics of hemogram indicators
Показатели гемограммы Hemogram Indicators | Клинико-гематологическое улучшение (отсутствие трансфузионной зависимости) Clinical and hematological improvements (no transfusion dependence) | Частичная ремиссия Partial Remission | Полная ремиссия Complete Remission |
|---|---|---|---|
Гемоглобин Hemoglobin | ≥80,0 г/л | ≥100 г/л ≥100 g/L | ≥120 г/л ≥120 g/L |
Гранулоциты Granulocytes | ≥1,0 * 10 9/л для нетяжелой формы АА ≥1,0 х 109/L for the non-heavy AA form ≥0,5 * 10 9/л для тяжелой и сверхтяжелой форм АА ≥0,5 х 109/L for severe and very severe AA forms | ≥1,5 * 10 9/л ≥1,5 х 10 9/L | ≥2 * 10 9/л ≥2 х 10 9/L |
Тромбоциты Platelets | ≥20 × 10 9/л | ≥80 × 10 9/л ≥80 × 10 9/L | ≥150 * 10 9/л ≥150 х 10 9/L |
Специальных методов реабилитации при АА не существует. Реабилитация при возникновении осложнений течения заболевания и лечения проводится в рамках соответствующих нозологий. Рекомендуется вести здоровый образ жизни, исключить избыточную инсоляцию и тепловые физиотерапевтические процедуры. Рекомендуется наблюдение гематолога: профилактические осмотры и контроль за анализами крови ежегодно.
Частота наблюдения больных АА после завершения лечения не регламентирована. В течение первого года после завершения лечения больной должен наблюдаться у гематолога не реже 1 раза в 3 месяца. Далее частота наблюдения устанавливается гематологом индивидуально, в зависимости от общего состояния и самочувствия пациента, осложнений проведенной терапии, достигнутого ответа на терапию, но не должна быть реже 1 раза в год. При диспансерном наблюдении кроме осмотра больного и сбора анамнеза и жалоб необходимо выполнять общий анализ крови с исследованием лейкоцитарной формулы. Остальные методы обследования могут применяться на усмотрение гематолога при наличии показаний.
Следует учесть, что у больного могут быть необычные проявления болезни, а также сочетание конкретной болезни с другими патологиями, что может диктовать лечащему врачу изменения в алгоритме выбора оптимальной тактики диагностики и лечения.

Приложение 1. Алгоритм диагностики и выбора метода лечения больных АА
Appendix 1. Diagnostic algorithm and treatment options for patients with AA

Приложение 2. Дифференциальная диагностика AA и апластических синдромов (вторичные аплазии костного мозга]
Appendix 2. Differential diagnosis of AA and aplastic syndromes (secondary bone marrow aplasia)

Приложение 3. Алгоритм лечения АА
Appendix 3. Algorithm for the treatment of AA
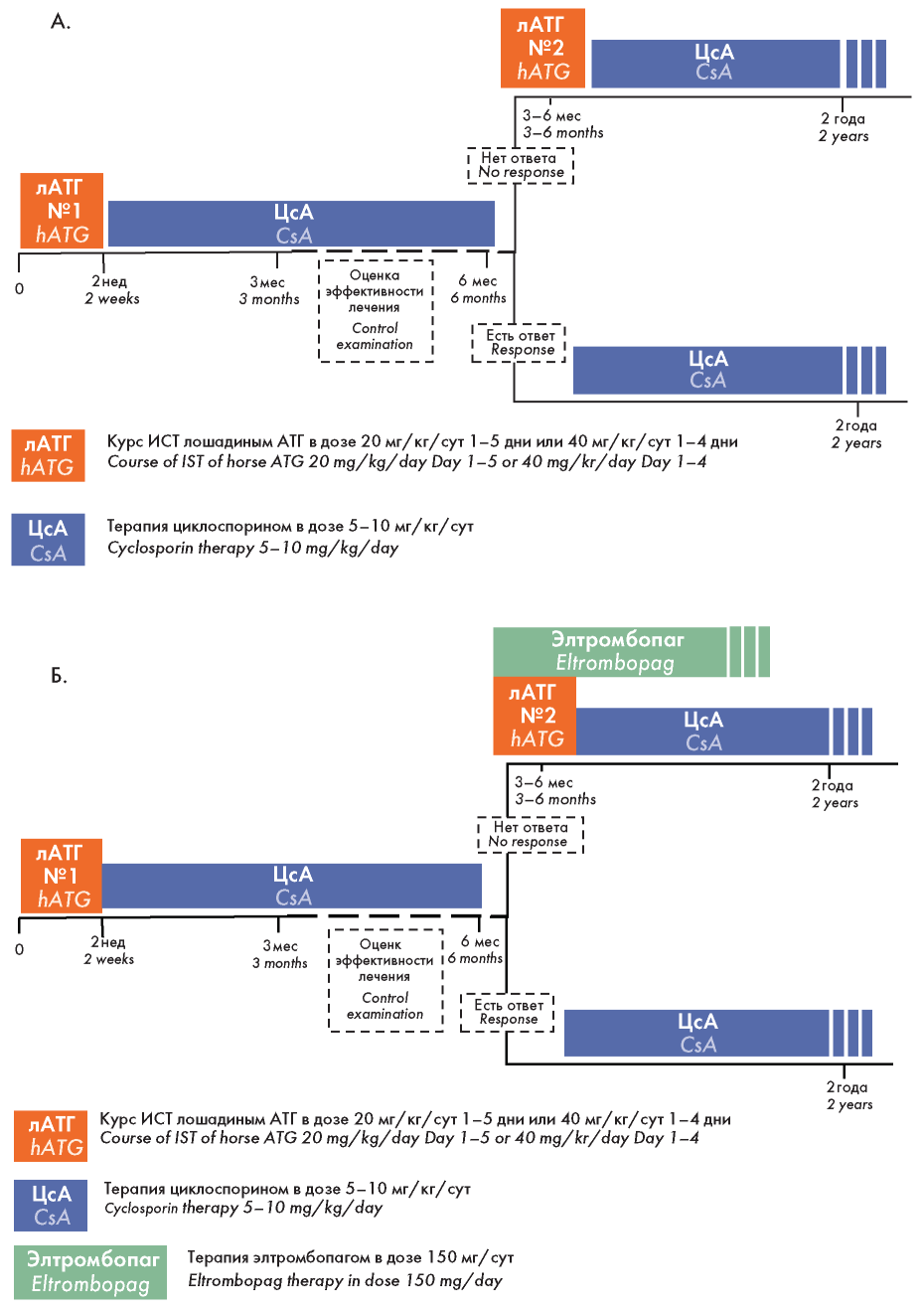
Приложение 4. Протоколы комбинированной ИСТ больных АА: А — классический протокол ИСТ; Б — протокол, включающий агонисты тромбопоэтиновых рецепторов у больных, рефрактерных к первому курсу АТГ; В — Протокол программы комбинированной ИСТ, включающей агонисты тромбопоэтиновых рецепторов в период индукции ремиссии и лечения рецидивов апластической анемии; Г — протокол комбинированной терапии, при наличии противопоказаний к терапии АТГ на первом этапе.
Appendix 4. Combined IST Protocols of AA patients: A — classic IST protocol; Б — protocol, including thrombopoietin receptor agonists in patients refractory to the 1st course of ATG; B — Protocol of the combined IST program, including thrombopoietin receptor agonists during the induction of remission and treatment of refractory of aplastic anemia; Г — protocol of combination therapy, in the presence of contraindications to ATG therapy in the first stage.

Приложение 5. Протокол применения АТГ при АА
Appendix 5. Protocol of ATG treatment of AA patients
Приложение 5. Протокол применения АТГ при АА
Appendix 5. Protocol of ATG treatment of AA patients
За 2 дня до начала терапии АТГ 2 days before the start of ATG therapy | |
|---|---|
Триметоприм/сульфаметоксазол Trimetoprim/sulfametoxasol | 480 мг внутрь 1 раз в сутки или 960 мг внутрь 3 раза в неделю в течение 3 недель 480 mg/day p. o. or 960 mg/day p. o. 3 times a week during 3 weeks |
Преднизолон Prednisolone | 30 мг/сут внутрь 30 mg/day p. o. |
Препараты кальция Calcium tablets | 1 -2 таблетки в день в течение 4 недель 1-2 tabs/day the course of 4 week |
Ежедневно в течение 5 дней Daily for the course of 5 days | |
АТГ 14 ATG14 | Лошадиный антитимоцитарный глобулин (Атгам), 20 мг/кг/сут 2 внутривенно капельно в течение 12 часов 3 20 mg/kg/d intravenous infusion (IV) for 12 hours |
Глюкокортикоидные гормоны Corticosteroids | Преднизолон 60 мг или метилпреднизолон 125-250 мг 2 раза в сутки внутривенно капельно (до и после АТГ) Prednisolone 60 mg or metilprednisolone 125-250 mg twice a day IV (before and after ATG) |
Антигистаминные препараты Antihistamines | 2 раза в сутки внутривенно капельно (до и после АТГ) twice a day IV (before and after ATG) |
Трансфузионная терапия Transfusion Therapy | |
Донорские тромбоциты Donor platelets | Концентрат тромбоцитов при концентрации тромбоцитов менее 20 * 10 9/л и/или при геморрагическом синдроме Platelet concentrate, if the platelet concentration is less than 20 x 109/L and/or in hemorrhagic syndrome |
Донорские эритроциты Donor red blood cells | Эритроцитная взвесь, при концентрации гемоглобина крови менее 80 г/л Erythrocyte concentrates below the Hb threshold 80 g/L |
Примечание. 1 — Перед первым введением АТГ проверяется индивидуальная чувствительность больного к белку с помощью двукратной (внутри- кожной и подкожной) пробы с препаратом АТГ (0,1 % раствор) или противостолбнячной сывороткой. 2 — При отсутствии тяжелых инфекционных осложнений в течение 2-3 месяцев перед началом ИСТ возможно использование 40 мг/кг в сутки в течение 4 дней. 3 — Суточная доза препарата вводится в 1200-1600 мл 0,9% раствора хлорида натрия. 4 — Первый день курса — день первого введения АТГ. С 14-го дня курса (при отсутствии или после исчезновения клинической картины сывороточной болезни) начинают постепенное уменьшение суточной дозы преднизолона, принимаемого внутрь, до полной отмены на 21-24-й день.
Note.1 — Individual sensitivity to the horse protein had been verified before infusion after intra- or subcutaneous test with 0.1 % ATG or tetanus vaccine.2 — Dosage regime 40 mg/kg/day can be used for 4 days in the absence of severe infectious complications. 3 — The daily dose of the ATG is administered in 1200-1600 ml of 0.9% sodium chloride solution. 4 — The first day of the course is the day of the first administration of ATG. From the 14th day of the course (in the absence or after the disappearance of serum disease), a gradual reduction in the daily dose of prednisone taken orally begins, until complete cancellation on the 21-24th day.
Приложение 6. Обобщенная таблица диагностических исследований
Appendix 6. Generalized table of diagnostic studies
№ | Название исследования Diagnostic tool | Характерные при АА изменения Characteristic changes in AA |
|---|---|---|
1 | Клинический анализ крови Peripheral blood analysis | Концентрация гемоглобина, количество нейтрофилов и тромбоцитов равномерно снижены. На ранних стадиях может развиваться изолированная цитопения, особенно тромбоцитопения. Количество лимфоцитов обычно не снижается. Наличие однолинейной цитопении требует дальнейшего изучения: необходимо исключить волосатоклеточный лейкоз или наследственную недостаточность костного мозга из-за мутации GATA2 (синдром Эмбергера) The concentration of hemoglobin, the number of neutrophils and platelets is evenly reduced. In the early stages, isolated cytopenia may develop, especially thrombocytopenia. The number of lymphocytes is usually not reduced. The presence of single-line cytopenia requires further study: it is necessary to exclude hairy cell leukemia or hereditary bone marrow failure due to the GATA2 mutation (Emberger syndrome) |
2 | Ретикулоциты Reticulocytes | Ретикулоцитопения менее 60 * 10 9/л при автоматическом подсчете Reticulocytopenia less than 60 х 109/L with automatic counting |
3 | Морфология клеток крови Blood cell morphology | Макроцитоз и анизопойкилоцитоз. Нейтрофилы могут быть с токсической зернистостью. Тромбоциты в основном небольшие по размеру. Необходимо исключить наличие диспластических нейтрофилов, патологических тромбоцитов, бластов и других патологических клеток, таких как «волосатые» клетки Macrocytosis and anisopoikilocytosis. Neutrophils may have toxic granularity. Platelets are mostly small in size. It is necessary to exclude the presence of dysplastic neutrophils, pathological platelets, blasts and other pathological cells, such as "hairy" cells |
4 | Тест на ломкость хромосом (с диэпоксибутаном — ДЭБ-тест) Chromosome fragility test (with diepoxybutane — DEB test) | Исключение анемии Фанкони Exclusion of Fanconi Anemia |
5 | Проточная цитометрия Flow cytometry | Определение процента ГФИ-дефектных клеток в периферической крови (ПНГ-клон) Determination of the percentage of GPI—defective cells in peripheral blood (PNH-clone) |
6 | Дефицит B12 и фолатов B12 and folate deficiency | Может встречаться у больных АА, однако аплазия костного мозга из-за дефицита витаминов встречается крайне редко May occur in patients with AA, but bone marrow aplasia due to vitamin deficiency is extremely rare |
7 | Печеночные пробы Liver function tests | Необходимо проводить с целью выявления гепатит-ассоциированной АА Must be performed to detect hepatitis-associated AA |
8 | Вирусные инфекции: гепатит A/B/C, Эпштейн — Барр вирус (ЭБВ), цитомегаловирус (ЦМВ), вирус иммунодефицита человека (ВИЧ) и парвовирус B19 Viral infections: hepatitis A/B/C, Ebstein-Barr virus, cytomegalovirus, human immunodeficiency virus (HIV) and parvovirus B19 | АА вследствие гепатита встречается редко, обычно возникает через 2-3 месяца после острого приступа гепатита и более часто встречается у молодых мужчин. При постгепатитной АА серология часто отрицательна. ЦМВ следует оценивать, если в дальнейшем рассматривается ТГСК. ВИЧ чаще вызывает изолированные цитопении, но является очень редкой причиной АА. Аналогичным образом, парвовирус В19 чаще ассоциируется с приобретенной красноклеточной аплазией AA due to hepatitis is rare, usually occurs 2-3 months after an acute attack of hepatitis and is more common in young men. In posthepatitis AA, serology is often negative. CMV should be assessed if further considered by the HSCT. HIV often causes isolated cytopenia, but is a very rare cause of AA. Similarly, parvovirus B19 is more often associated with acquired red cell aplasia |
9 | Антинуклеарный фактор и антитела к двуспиральной ДНК Antinuclear factor and antibodies to double-stranded DNA | Панцитопения при системной красной волчанке может быть аутоиммунной с клеточным костным мозгом, ассоциироваться с миелофиброзом или редко — с гипоклеточным костным мозгом Pancytopenia in systemic lupus erythematosus may be autoimmune with cell bone marrow, associated with myelofibrosis, or rarely with hypoccellular bone marrow |
10 | Определение длины теломерных участков ДНК в лейкоцитах периферической крови* Determination of telomer length in peripheral blood leukocytes | Скрининг заболеваний на мутации генов теломеразного комплекса при врожденном дискератозе, менее специфичен для взрослых с АА с мутациями TERC/TERT; короткие приобретенные теломеры могут также возникать при приобретенной AA с уменьшенным пулом стволовых клеток Screening for telomerase complex gene mutations in congenital dyskeratosis is less specific for adults with AA with TERC/TERT mutations; short acquired telomeres can also occur with acquired AA with a reduced stem cell pool |
11 | Секвенирование нового поколения* Next generation sequencing (NGS) | • Мутации в генах теломеразного комплекса • Другие врожденные АА • Приобретенные соматические мутации, типичные для злокачественных миелоидных новообразований, для дифференциальной диагностики АА и гипоклеточного МДС, для раннего выявления клональной эволюции в МДС/ОМЛ • Mutations in the genes of the telomerase complex • Other congenital AA • Acquired somatic mutations, typical for malignant myeloid neoplasms, for the differential diagnosis of AA and hypocellular MDS, for early detection of clonal evolution in MDS/AML |
Примечание. * — Дополнительные методы диагностики, не являющиеся обязательными.
Note. * — additional diagnostic methods that are not required.
1. Young N.S. Pathophysiologic mechanisms in acquired aplastic anemia. Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program. 2006. P. 72–7.
2. Zeng Y., Katsanis E. The complex pathophysiology of acquired aplastic anaemia. Clin Exp Immunol. 2015; 180(3): 361–70. DOI: 10.1111/cei.12605.
3. Medinger M., Drexler B., Lengerke C., et al. Pathogenesis of acquired aplastic anemia and the role of the bone marrow microenvironment. Front Oncol. 2018; 8: 1–10. DOI: 10.3389/fonc.2018.00587.
4. Isidori A., Borin L., Elli E., et al. Iron toxicity — Its effect on the bone marrow. Blood Rev. 2018; 32(6): 473–9. DOI: 10.1016/j.blre.2018.04.004.
5. Marsh J.C.W., Kulasekararaj A.G. Management of the refractory aplastic anemia patient: what are the options? Blood. 2013; 122(22): 3561–7. DOI: 10.1182/blood-2013-05-498279.
6. Frickhofen N., Heimpel H., Kaltwasser J.P., et al. Antithymocyte globulin with or without cyclosporin A: 11-Year follow-up of a randomized trial comparing treatments of aplastic anemia. Blood. 2003; 101(4): 1236–42. DOI: 10.1182/blood-2002-04-1134.
7. Afable M.G., Tiu R. V, Maciejewski J.P. Clonal evolution in aplastic anemia. Hematology Am Soc Hematol Educ Program. 2011; 2011: 90–5. DOI: 10.1182/asheducation-2011.1.90.
8. Pu J.J., Mukhina G., Wang H., et al. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol. 2011; 87(1): 37–45. DOI: 10.1111/j.1600-0609.2011.01615.x.
9. Socié G., Mary J.-Y., de Gramont A., et al. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. Lancet. 1996; 348(9027): 573–7. DOI: 10.1016/S0140-6736(95)12360-1.
10. Kulagin A., Lisukov I., Ivanova M., et al. Prognostic value of paroxysmal nocturnal haemoglobinuria clone presence in aplastic anaemia patients treated with combined immunosuppression: Results of two-centre prospective study. Br J Haematol. 2014; 164(4): 546–54. DOI: 10.1111/bjh.12661.
11. Li Y., Li X., Ge M., et al. Long-term follow-up of clonal evolutions in 802 aplastic anemia patients: A single-center experience. Ann Hematol. 2011; 90(5): 529– 37. DOI: 10.1007/s00277-010-1140-9.
12. Кулагин А.Д., Лисуков И.А., Птушкин В.В. и др. Национальные рекомендации по диагностике и лечению пароксизмальной ночной гемоглобинурии. Онкогематология. 2014; 2: 20–8.
13. Kaufman D.W., Kelly J.P., Issaragrisil S., et al. Relative incidence of agranulocytosis and aplastic anemia. Am J Hematol. 2006; 81: 65–67. DOI: 10.1002/ajh.20489.14.
14. Михайлова Е.А. Протокол программного лечения больных апластической анемией: комбинированная иммуносупрессивная терапия. В кн. под ред. В.Г. Савченко. Программное лечение заболеваний системы крови. М.: Практика; 2012: 135–50.
15. Killick S.B., Bown N., Cavenagh J., et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016; 172(2): 187–207. DOI: 10.1111/bjh.13853.
16. Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011; 2011: 480–6. DOI: 10.1182/asheducation-2011.1.480.
17. Soulier J. Fanconi anemia. Hematology Am Soc Hematol Educ Program. 2011; 2011: 492–7. DOI: 10.1182/asheducation-2011.1.492.
18. Marsh J.C.W., Ball S.E., Cavenagh J., et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009; 147(1): 43–70. DOI: 10.1111/j.1365-2141.2009.07842.x.
19. Peslak S.A., Olson T., Babushok D.V. Diagnosis and Treatment of Aplastic Anemia. Vol. 18, Current Treatment Options in Oncology. Springer New York LLC; 2017. DOI: 10.1007/s11864-017-0511-z.
20. Barone A., Lucarelli A., Onofrillo D., et al. Diagnosis and management of acquired aplastic anemia in childhood. Guidelines from the Marrow Failure Study Group of the Pediatric Haemato-Oncology Italian Association (AIEOP). Blood Cells, Mol Dis. 2015; 55(1): 40–7. DOI: 10.1016/j.bcmd.2015.03.007.
21. Frisch B., Lewis S.M. The bone marrow in aplastic anaemia: diagnostic and prognostic features. J Clin Pathol. 1974; 27(3): 231–41. DOI: 10.1136/jcp.27.3.231.
22. Maciejewski J.P., Mufti G.J. Whole genome scanning as a cytogenetic tool in hematologic malignancies. Blood. 2008; 112(4): 965–74. DOI: 10.1182/blood-2008-02-130435.
23. Maciejewski J.P., Risitano A., Sloand E.M., et al. Distinct clinical outcomes for cytogenetic abnormalities evolving from aplastic anemia. Blood. 2002; 99(9): 3129–35. DOI: 10.1182/blood.V99.9.3129.
24. Gupta V., Brooker C., Tooze J.A., et al. Clinical relevance of cytogenetic abnormalities at diagnosis of acquired aplastic anaemia in adults. Br J Haematol. 2006; 134(1): 95–9. DOI: 10.1111/j.1365-2141.2006.06105.x.
25. Ольшанская Ю.В. Михайлова Е.А. Домрачева Е.В. и др. Клональные хромосомные перестройки у больных апластической анемией в начале заболевания и при трансформации. Терапевтический архив. 2006; 78(7): 31–7.
26. Dumitriu B., Feng X., Townsley D.M., et al. Red cells, iron, and erythropoiesis: Telomere attrition and candidate gene mutations preceding monosomy 7 in aplastic anemia. Blood. 2015; 125(4): 706–9. DOI: 10.1182/blood-2014-10-607572.
27. Jerez A., Clemente M.J., Makishima H., et al. STAT3 mutations indicate the presence of subclinical T-cell clones in a subset of aplastic anemia and myelodysplastic syndrome patients. Blood. 2013; 122(14): 2453–9. DOI: 10.1182/blood-2013-04-494930.
28. Du Y., Long Z., Chen M., et al. Observational Monitoring of Patients with Aplastic Anemia and Low/Intermediate-1 Risk of Myelodysplastic Syndromes Complicated with Iron Overload. Acta Haematol. 2017; 138(2): 119–28. DOI: 10.1159/000479422.
29. Borowitz M.J., Craig F.E., Digiuseppe J.A., et al. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by fl ow cytometry. Cytom Part B — Clin Cytom. 2010; 78(4): 211–30. DOI: 10.1002/cyto.b.20525.
30. Ogawa S. Clonal hematopoiesis in acquired aplastic anemia. Blood. 2016; 3: 337–47. DOI: 10.1182/blood-2016-01-636381.
31. Parker C.J. Update on the diagnosis and management of paroxysmal nocturnal hemoglobinuria. Hematol Am Soc Hematol Educ Progr. 2016; 2016(1): 208–16. DOI: 10.1182/asheducation-2016.1.208.
32. Alter B.P. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program. 2007; 29–39. DOI: 10.1182/asheducation-2007.1.29.
33. Михайлова Е.А., Фидарова З.Т., Устинова Е.Н. и др. Комбинированная иммуносупрессивная терапия больных апластической анемией: повторные курсы антитимоцитарного глобулина. Гематология и трансфузиология. 2014; 59(4): 11–8.
34. Young N.S., Bacigalupo A., Marsh J.C.W. Aplastic Anemia: Pathophysiology and Treatment. Biol Blood Marrow Transplant. 2010; 16(1): S119–25. DOI: 10.1016/j.bbmt.2009.09.013.
35. Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017; 129(11): 1428–36. DOI: 10.1182/blood-2016-08-693481.
36. Scheinberg P., Young N.S. How I treat acquired aplastic anemia. Blood. 2012; 120(6): 1185–96. DOI: 10.1182/blood-2011-12-274019.
37. Desmond R., Townsley D.M., Dumitriu B., et al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood. 2014; 123(12): 1818–25. DOI: 10.1182/blood-2013-10-534743.
38. Lee J.W., Yoon S.-S., Shen Z.X., et al. Iron chelation therapy with deferasirox in patients with aplastic anemia: A subgroup analysis of 116 patients from the EPIC trial. Blood. 2010; 116(14): 2448–54. DOI: 10.1182/blood-2010-01-261289.
39. Kelsey P., Murphy M.F., Brown M., et al. Guidelines for the use of platelet transfusions. Br J Haematol. 2003; 122(1): 10–23. DOI: 10.1046/j.1365-2141.2003.04468.x.
40. Bacigalupo A., Brand R., Oneto R., et al. Treatment of acquired severe aplastic anemia: Bone marrow transplantation compared with immunosuppressive therapy — The European Group for blood and marrow transplantation experience. Semin Hematol. 2000; 37(1): 69–80.
41. Bacigalupo A., Würsch A., Hows J.M., et al. Long‐term follow‐up of severe aplastic anaemia patients treated with antithymocyte globulin. Br J Haematol. 1989; 73(1): 121–6. DOI: 10.1111/j.1365-2141.1989.tb00230.x.
42. Rosenfeld S., Follmann D., Nunez O., et al. Antithymocyte Globulin and Cyclosporine for Severe Aplastic Anemia: Association between Hematologic Response and Long-term Outcome. J Am Med Assoc. 2003; 289(9): 1130–5. DOI: 10.1001/jama.289.9.1130.
43. Marchiò C., Dowsett M., Reis-Filho J.S. Revisiting the technical validation of tumour biomarker assays: How to open a Pandora’s box. BMC Med. 2011; 9. DOI: 10.1186/1741-7015-9-41.
44. Bacigalupo A., Hows J., Gluckman E., et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): A report of the EBMT SAA Working Party. Br J Haematol. 1988; 70(2): 177–82.
45. Scheinberg P., Townsley D., Dumitriu B., et al. Horse antithymocyte globulin as salvage therapy after rabbit antithymocyte globulin for severe aplastic anemia. Am J Hematol. 2014; 89(5): 467–9. DOI: 10.1002/ajh.23669.
46. Scheinberg P. Aplastic anemia: therapeutic updates in immunosuppression and transplantation. Hematology Am Soc Hematol Educ Program. 2012; 2012: 292–300. DOI: 10.1182/asheducation-2012.1.292.
47. Miano M., Dufour C. The diagnosis and treatment of aplastic anemia: a review. Int J Hematol. 2015; 101(6): 527–35. DOI: 10.1007/s12185-015-1787-z.
48. Peffault de Latour R., Tabrizi R., Marcais A., et al. Nationwide survey on the use of horse antithymocyte globulins (ATGAM) in patients with acquired aplastic anemia: A report on behalf of the French Reference Center for Aplastic Anemia. Am J Hematol. 2018; 93(5): 635–42. DOI: 10.1002/ajh.25050.
49. Valdez J.M.J.M., Scheinberg P., Nunez O., et al. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011; 52(6): 726–35. DOI: 10.1093/cid/ciq245.
50. Winkler T., Fan X., Cooper J., et al. Treatment optimization and genomic outcomes in refractory severe aplastic anemia treated with eltrombopag. Blood. 2019; 133(24): 2575–85. DOI: 10.1182/blood.2019000478.
51. Olnes M.J., Scheinberg P., Calvo K.R., et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med. 2012; 367(1): 11–9. DOI: 10.1056/NEJMoa1200931.
52. Desmond R., Townsley D.M., Dunbar C., et al. Eltrombopag in Aplastic Anemia. Seminars in Hematology. 2015. DOI: 10.1053/j.seminhematol.2014.10.002.
Михайлова Елена Алексеевна, доктор медицинских наук, профессор, ведущий научный сотрудник отделения высокодозной интенсивной химиотерапии гемобластозов и депрессий кроветворения с круглосуточным стационаром
тел.: +7(495) 613-26-90
Фидарова Залина Таймуразовна*, кандидат медицинских наук, заведующая отделением химиотерапии гемобластозов и депрессий кроветворения с дневным стационаром
тел.: +7(495) 612-45-92
Троицкая Вера Витальевна, кандидат медицинских наук, заведующая отделением интенсивной высокодозной химиотерапии гемобластозов и депрессий кроветворения с круглосуточным и дневным стационарами
тел.: +7(495) 612-45-92
Кулагин Александр Дмитриевич, доктор медицинских наук, профессор кафедры гематологии, трансфузиологии и трансплантологии
тел.: +7 (812) 338-62-84
Михайлова Е.А., Фидарова З.Т., Троицкая В.В., Клясова Г.А., Кулагин А.Д., Воронова Е.В., Двирнык В.Н., Гальцева И.В., Ковригина А.М., Обухова Т.Н., Гапонова Т.В., Паровичникова Е.Н., Савченко В.Г. Клинические рекомендации по диагностике и лечению апластической анемии (редакция 2019 г.). Гематология и трансфузиология. 2020;65(2):208-226. https://doi.org/10.35754/0234-5730-2020-65-2-208-226
Mihailova E.A., Fidarova Z.T., Troitskaya V.V., Klyasova G.A., Kulagin A.D., Voronova E.V., Dvirnyk V.N., Galtseva I.V., Kovrigina A.M., Obukhova T.N., Gapanova T.V., Parovichnikova E.N., Savchenko V.G. Clinical recommendations for the diagnosis and treatment of aplastic anemia (2019 edition). Russian journal of hematology and transfusiology. 2020;65(2):208-226. (In Russ.) https://doi.org/10.35754/0234-5730-2020-65-2-208-226
![]()
125167, Москва, Новый Зыковский проезд, 4
ФГБУ «НМИЦ гематологии» Минздрава России
тел.: 8-926-816-3887
e-mail: o.levchenko@htjournal.ru






































