


Содержание
Перейти к:
https://doi.org/10.35754/0234-5730-2022-67-4-570-578
Перейти к:
Введение. Наиболее распространенным представлением о функции фактора свертывания крови XII (FXII) является его участие во внутреннем пути свертывания крови. Однако биологическая роль FXII многообразна.
Цель – обзор разнообразных биологических функций FXII.
Основные сведения. FXII является сериновой протеазой. Структура FXII имеет высокую степень гомологии с плазминогеном, тканевым активатором плазминогена и урокиназным активатором плазминогена. Активированный FXII (FXIIa) имеет пять субстратов: высокомолекулярный кининоген, прекалликреин, FXI, плазминоген, белки комплемента (C1s, С1r). FXII обеспечивает гемостатическое равновесие, участвуя в процессах свертывания крови и фибринолиза. FXII регулирует воспалительные и аллергические реакции, взаимодействуя с калликреин-кининовой системой и системой комплемента. FXII имеет биологическую активность в различных клетках in vivo: эндотелиоцитах, тромбоцитах, моноцитах, нейтрофилах, фибробластах, дендритных клетках, что определяет его многообразную роль в физиологических и патологических процессах.
Яковлева Е.В., Зозуля Н.И. Физиологическая и патологическая роль фактора свертывания крови XII. Гематология и трансфузиология. 2022;67(4):570-578. https://doi.org/10.35754/0234-5730-2022-67-4-570-578
Yakovleva E.V., Zozulya N.I. Physiological and pathological role of factor XII. Russian journal of hematology and transfusiology. 2022;67(4):570-578. (In Russ.) https://doi.org/10.35754/0234-5730-2022-67-4-570-578
Наиболее распространенным знанием о функции фактора свертывании крови XII (FXII), или фактора Хагемана, является его участие во внутреннем пути свертывания крови. Однако биологическая роль FXII многообразна.
Цель настоящей работы — продемонстрировать разнообразие биологических функций FXII.
Образование фибрина может быть вызвано двумя путями: повреждением сосудистой стенки (внешний путь свертывания крови) или контактной активацией с вовлечением отрицательно заряженных поверхностей (внутренний путь свертывания крови). Внутренний путь свертывания инициируется FXII c участием высокомолекулярного кининогена (ВМК) и прекалликреина (ПК), что приводит к активации фактора свертывания крови XI (FXI) и далее формированию теназного комплекса.
FXII по своим биохимическим свойствам, как и большинство факторов свертывания крови, является протеолитическим белком, сериновой протеазой. FXII представляет собой гликопротеин, имеет средний молекулярный вес (80 кДа) по сравнению с другими факторами свертывания крови. Концентрация в плазме составляет ~40 мкг/мл (~500 нмоль/л); нормальная активность — 70–150 % [1–3]. Во время беременности активность FXII в плазме увеличивается [4]. Период полужизни FXII составляет 50–70 часов. FXII синтезируется в печени, однако он также образуется и в лейкоцитах [5–7]. Ген F12 локализован на длинном плече 5-й хромосомы (5q33), включает 12 килобаз и состоит из 14 экзонов и 13 интронов [5][8–10]. Ген кодирует последовательность 596 аминокислот.
Начиная с N-конца, в структуре FXII имеется лидер-пептид, далее следуют домен II типа фибронектина, домен ростового фактора (EGF-подобный домен), домен I типа фибронектина, снова EGF-подобный домен, крингл-домен, домен богатый пролином, каталитический домен (рис. 1) [4][11][12]. У каждого домена своя функция. Домен II типа фибронектина обеспечивает взаимодействие с FXI, с цинком, с искусственной поверхностью [5][13–17]. Как следует из названия домена, его структура гомологична последовательности аминокислотных остатков фибронектина [18]. FXII взаимодействует с эндотелиальными клетками через рецептор урокиназного активатора плазминогена (urokinase-type plasminogen activator, uPA), цитокератина I, gC1qR, тромбоцитами через комплекс GPIba-IX-V, нейтрофилами [19–24]. Однако это взаимодействие возможно в присутствии достаточных концентраций ионов цинка [20]. In vivo источником ионов цинка являются активированные тромбоциты. Связывание с ионами цинка приводит к конформационным изменениям в структуре FXII, что обеспечивает его взаимодействие с указанными клетками. Две области FXII гомологичны аминокислотной последовательности эпидермального фактора роста (epidermal growth factor, EGF). Такая гомология также определяется в трансформирующем факторе роста (transforming growth factor, TGF) типа 1, тканевом активаторе плазминогена (tissue plasminogen activator, tPA), одноцепочечном урокиназном активаторе плазминогена и некоторых факторах свертывания крови, например FX [5][11]. EGF является митогеном для различных клеток и стимулирует плейотропный ответ в клетках-мишенях [25][26]. Неизвестно, опосредуют ли EGF-подобные домены FXII эти действия. Два EGF-подобных домена, наряду с доменом II типа фибронектина, имеют сайты связывания с ионами цинка [24]. Домен I типа фибронектина — небольшой домен, включающий 43 аминокислотных остатка, разделяющих 2 EGF-домена. Предполагается, что домен I типа фибронектина обеспечивает взаимодействие с искусственной поверхностью, фибрином и гепарином [11][27]. Крингл-домен включает 80 аминокислотных остатков и гомологичен tPA, протромбину [5][11]. Он обеспечивает взаимодействие с искусственной поверхностью, фибриногеном, фибрином [4][5][11][12]. Далее следует пролин-богатый домен, в котором 33 % аминокислот представлены пролином. Этот домен не имеет какой-либо гомологии с другими белками. Значение этого домена остается неопределенным [11]. Каталитический домен является глобулярным. Расщепление связи Arg353-Val354 превращает одноцепочечный зимоген FXII в активированную форму — α-FXIIa [5]. In vivo этот расщепленный белок циркулирует в виде двухцепочечного белка, тяжелой цепи ~50 кДа (353 аминокислотных остатка) и легкой цепи ~30 кДа (243 аминокислотных остатка), удерживаемых вместе дисульфидной связью. Восстановление дисульфидной связи высвобождает легкую цепь ~30 кДа в виде β-FXIIa [4][11][12]. β-FXIIa сохраняет свою протеолитическую активность по отношению к белковым субстратам, но не способен связываться с отрицательно заряженными поверхностями и, следовательно, способствовать свертыванию крови [4][11][28].
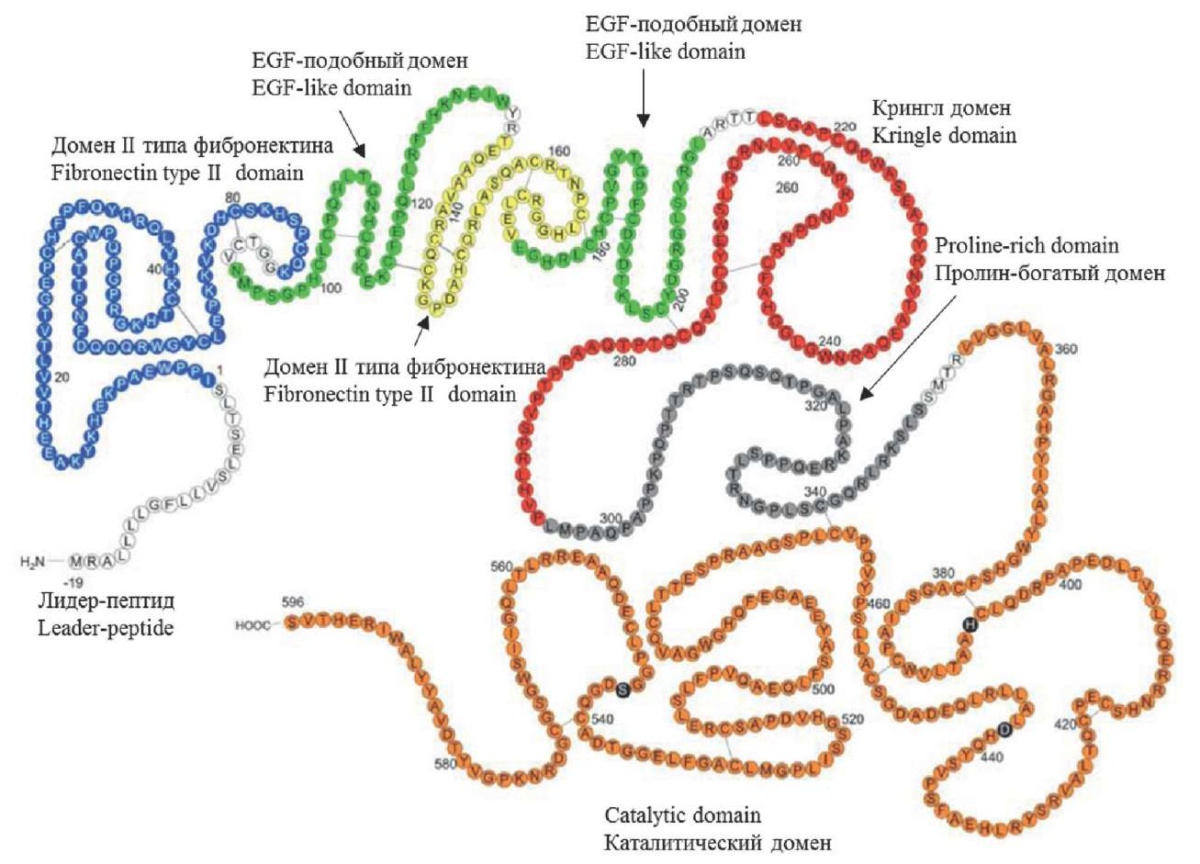
Рисунок 1. Структура FXII [11]
Figure 1. FXII structure [11]
FXIIa активирует свертывание крови по внутреннему пути. Контактная активация на нефизиологических поверхностях — защитный механизм. Примером активации свертывающей системы по внутреннему пути in vitro являются лабораторные коагуляционные тесты, а in vivo — соприкосновение крови с полимерными поверхностями при катетеризации, проведении диализа, экстракорпоральной мембранной оксигенации, сердечно-легочном шунтировании, установке искусственных клапанов сердца. Более того, биологическими субстанциями, приводящими к контактной активации, являются ДНК, РНК, денатурированные белки (например, β-амилоид), открытый коллаген стенки сосуда, полифосфат тромбоцитов ивнеклеточные ловушки (сетки) нейтрофильных клеток, которые служат платформой для аутоактивации FXII. Это, в свою очередь, лежит в основе патофизиологии инфекционных заболеваний или онкологических процессов [3][12]. FXII обладает уникальной способностью аутоактивации, однако это событие еще не до конца изучено. Процесс аутоактивации FXII очень медленный, происходит в течение 90–120 мин. при контакте с отрицательно заряженной поверхностью и называется твердофазной активацией. Процесс активации FXII значительно ускоряется в присутствии ВМК, прекалликреина и называется активацией в жидкой фазе [3][11][28][29]. FXIIa активирует прекалликреин в плазменный калликреин, это приводит креципрокной (взаимной) активации FXII. Последующий процесс амплификации заключается в нарастающей активации прекалликреина и FXIIa [3]. ВМК принимает участие в контактной активации как неэнзиматический кофактор, который имеет сайты связывания анионных поверхностей, прекалликреина, FXI. FXIIa и калликреин расщепляют ВМК, тем самым высвобождается брадикинин. FXIIa активирует FXI, запуская коагуляционный каскад и образование тромбина с последующей независимой от FXII активацией им FXI. Таким образом, контактная система активации включает четыре белка и характеризуется процессами аутоактивации, реципрокной активацией и амплификацией FXIIa. Молекулярный комплекс FXII, FXI, прекалликреин, ВМК является связующим звеном коагуляции и воспаления (рис. 2) [30][31]. Контактная система активации находится в тесной взаимосвязи с калликреин-кининовой системой, которая инициируется образованием плазменного калликреина. Плазменный калликреин, FXIIa расщепляют ВМК с образованием брадикинина и фрагментов кининогена. Брадикинин, взаимодействуя с G-белком, рецепторами В2 и В1, влияет на сосудистые реакции. Он является непосредственным участником воспалительного процесса — расширяет сосуды, влияет на проницаемость сосудистой стенки, артериальное давление [3][31]. FXIIa активирует классический путь системы комплемента, активируя С1r и С1s. Нарушения системы контактной активации, активности калликреин-кининовой системы являются патогенетическим механизмом развития острых приступов наследственного ангионевротического отека. Одним из этиологических вариантов наследственного ангионевротического отека являются генетические дефекты FXII [3][31–35].
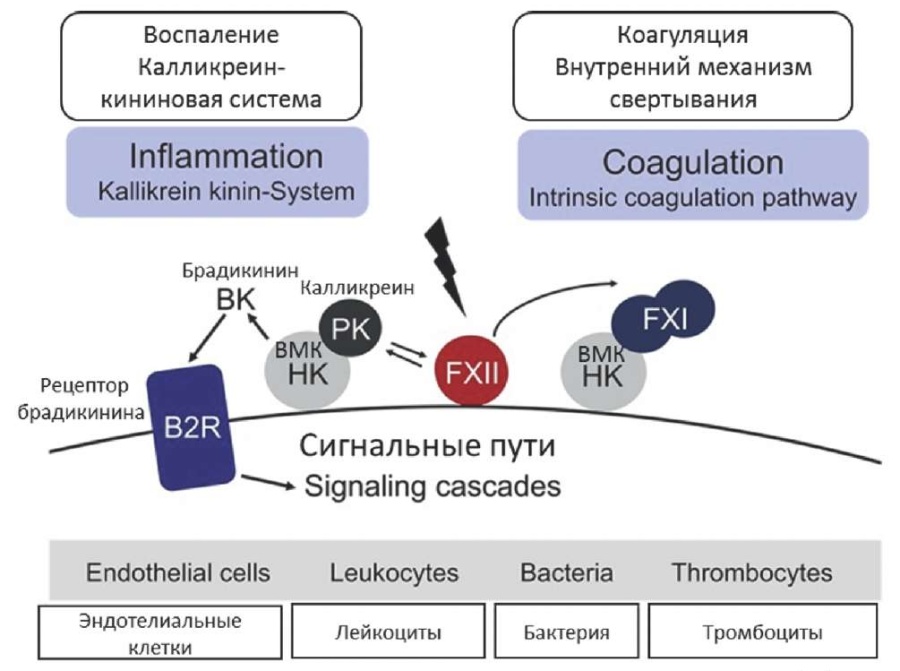
Рисунок 2. Контактная активация: взаимосвязь коагуляции и калликреин-кининовой системы [31]
Примечание: ВМК — высокомолекулярный кининоген.
Figure 2. Contact activation: relationship between coagulation and kallikrein-kinin system [31]
Note: HK — high molecular weight kininogen, PK — plasma kallikrein, B2R — bradykinin B2receptor, BK — bradykinin.
Структура FXII имеет высокую степень гомологии с плазминогеном, tPA, uPA. Контактная активация является инициатором внутреннего пути фибринолиза и повышает фибринолитическую активность плазмы [4]. Калликреин, FXIIa, FXIa способны расщеплять плазминоген, но менее эффективно, чем tPA и uPA [36–39]. Фибринолитическая активность FXIIa определяется тестом FXIIa-зависимого фибринолиза, принцип которого основан на выделении из плазмы крови эуглобулиновой фракции, содержащей плазминоген, фибриноген, факторы свертывания и не содержащей ингибиторов фибринолиза. При добавлении к этой фракции хлористого кальция образуется сгусток фибрина, который затем лизируется плазмином. Реакция активируется фактором XIIа [40].
Исследования активности калликреина in vitro показали, что он является активатором одноцепочечной урокиназы преимущественно на поверхности тромбоцитов и эндотелиальных клеток [41][42]. Одноцепочечный uPA (предшественник uPA) способен непосредственно активировать плазминоген c образованием плазмина, а затем плазмин превращает одноцепочечную форму в двухцепочечную, фибринолитическая активность которой в 2,5 раза выше [43][44]. Высвобождаемый при активации калликреина и FXII брадикинин в свою очередь является мощным и селективным индуктором высвобождения tPA из эндотелиальных клеток, что было продемонстрировано на человеческих и животных экспериментальных моделях [45][46]. Таким образом, FXIIa имеет пять субстратов: ВМК, прекалликреин, FXI, плазминоген, белки комплемента (C1s, С1r) [2][3].
Основным плазменным ингибитором протеаз α-FXIIa и β-FXIIa является ингибитор эстеразы С1 (ингибитор С1 или C1-ING). Ингибитор С1 связывает оба этих фермента необратимо и инактивирует их. Но когда FXIIa связан с отрицательно заряженной поверхностью, он защищен от инактивации ингибитором С1. Антитромбин III и ингибитор активатора плазминогена I (Plasminogen activator inhibitor-1, PAI-1), α-1-антитрипсин, α-2-макроглобулин, α-2-антиплазмин оказывают некоторое ингибирующее действие на FXIIa [47–53].
Зимоген FXII имеет биологическую активность в различных клетках in vivo. Первыми, описавшими влияние FXII на клеточную биологию, были Р. Chien и соавт., согласно данным которых FXIIa регулирует экспрессию рецепторов Fc гамма R1 моноцитов, а именно повышает ее [54]. В других исследованиях [26][55] показано, что FXII и FXIIa стимулировали митогенез с помощью митоген-активируемой протеинкиназы в клетках HepG2 (клеточная линия гепатоцеллюлярной карциномы человека).
В настоящее время имеется много данных, подтверждающих влияние FXII на васкулярную биологию. В эндотелии FXII, связываясь с рецептором активатора плазминогена урокиназного типа (uPAR) и далее через β1 интегрины и рецептор эпидермального фактора роста (EGFR), стимулирует рост, пролиферацию, ангиогенез после повреждения [56][57].
В нейтрофилах FXII также связывается с uPAR, что приводит к стимуляции Akt2 (фермент протеинкиназа, кодируемый геном Аkt2, влияет на накопление метаболитов и является участником сигнальных путей) и инициации нейтрофилами адгезии, миграции, хемотаксиса и в итоге к нетозу (программированной клеточной гибели нейтрофилов) [7][58].
В фибробластах TGF-β повышает экспрессию FXII, что приводит к их пролиферации, способствуя фиброзу тканей [57][59].
При исследованиях дендритных клеток установлено, что FXII играет роль в нейровоспалении. У больных рассеянным склерозом наблюдались высокие концентрации циркулирующего в крови FXII во время рецидива заболевания. В экспериментальных моделях рассеянного склероза на мышах показано уменьшение нейровоспаления у мышей F12-/- [60].
R. Hess и соавт. [61] описали регуляцию воспалительных процессов в легочной ткани, опосредованную FXII. Исследование включало 54 больных с острым респираторным дистресс-синдромом (ОРДС) и контрольную группу, состоявшую из43 человек. В жидкости бронхоальвеолярного лаважа у больных с ОРДС определялась высокая активность FXII, которая ассоциировались с высокой концентрацией интерлейкинов (ИЛ)-8, ИЛ-1β, ИЛ-6, фактора некроза опухоли альфа, и это, всвою очередь, коррелировало с выживаемостью. Имеются данные, что при идиопатическом легочном фиброзе, COVID-19, пневмонии повышается экспрессия и локальная активация FXII в легких. В 2015 г. опубликована работа, где впервые рассматривалась роль FXII в развитии легочного фиброза. Экспрессию FXII исследовали в легких больных идиопатическим легочным фиброзом. Была показана независимая от коагуляции профибротическая функция FXII, продуцируемого фибробластами легких. Генетическая абляция или фармакологическая блокада амидолитической активности FXII ингибитором кукурузного трипсина или инфестином-4 отменяли фиброгенез в модели повреждения легких блеомицином у мышей [62–65].
Таким образом, биологическая роль FXII многообразна. Он является участником двух противоположных процессов, обеспечивающих гемостатический баланс — внутреннего пути свертывания крови и фибринолиза. FXII запускает контактную систему активации, калликреин-кининовую систему, активирует систему комплемента. Тем самым FXII является связующим звеном коагуляции крови, воспалительных и аллергических реакций. Доказана его разнообразная роль в клеточном взаимодействии и клеточной регуляции, а именно в процессах пролиферации, ангиогенеза, клеточной миграции, экспрессии медиаторов воспаления. В связи с этим целесообразно учитывать роль FXII при различных патологических состояниях.
1. Revak S.D., Cochrane C.G., Johnston A.R., Hugli T.E. Structural changes accompanying enzymatic activation of human Hageman factor. J Clin Invest. 1974; 54(3): 619–27. DOI: 10.1172/JCI107799.
2. Saito H., Ratnoff O.D., Pensky J. Radioimmunoassay of human Hageman factor (factor XII). J Lab Clin Med. 1976; 88(3): 506–14.
3. Schmaier A.H., Stavrou E.X. Factor XII — What’s important but not commonly thought about. Res Pract Thromb Haemost. 2019; 3(4): 599–606. DOI: 10.1002/rth2.12235.
4. Colman R.W., Schmaier A.H. Contact system: A vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood. 1997; 90(10): 3819–43.
5. Cool D.E., Edgell C.J., Louie G.V., et al. Characterization of human blood coagulation factor XII cDNA. Prediction of the primary structure of factor XII and the tertiary structure of beta-factor XIIa. J Biol Chem. 1985; 260(25): 13666–76.
6. Gordon E.M., Gallagher C.A., Johnson T.R., et al. Hepatocytes express blood coagulation factor XII (Hageman factor). J Lab Clin Med. 1990; 115(4): 463–9.
7. Stavrou E.X., Fang C., Bane K.L., et al. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J Clin Invest. 2018; 128(3): 944–59. DOI: 10.1172/JCI92880.
8. Citarella F., Tripodi M., Fantoni A., et al. Assignment of human coagulation factor XII (fXII) to chromosome 5 by cDNA hybridization to DNA from somatic cell hybrids. Hum Genet. 1988; 80(4): 397–8. DOI: 10.1007/BF00273661.
9. Royle N.J., Nigli M., Cool D., et al. Structural gene encoding human factor XII is located at 5q33-qter. Somat Cell Mol Genet. 1988; 14(2): 217–21. DOI: 10.1007/BF01534407.
10. Cool D.E., MacGillivray R.T. Characterization of the human blood coagulation factor XII gene. Intron/exon gene organization and analysis of the 5’-flanking region. J Biol Chem. 1987; 262(28): 13662–73.
11. Stavrou E., Schmaier A.H. Factor XII: What does it contribute to our understanding of the physiology and pathophysiology of hemostasis & thrombosis. Thromb Res. 2010; 125(3): 210–5. DOI: 10.1016/j.thromres.2009.11.028.
12. Didiasova M., Wujak L., Schaefer L., Wygrecka M. Factor XII in coagulation, inflammation and beyond. Cell Signal. 2018; 51: 257–65. DOI: 10.1016/j.cellsig.2018.08.006.
13. Clarke B.J., Côté H.C., Cool D.E., et al. Mapping of a putative surface-binding site of human coagulation factor XII. J Biol Chem. 1989; 264(19): 11497–502.
14. Samuel M., Samuel E., Villanueva G.B. Histidine residues are essential for the surface binding and autoactivation of human coagulation factor XII. Biochem Biophys Res Commun. 1993; 191(1): 110–7. DOI: 10.1006/bbrc.1993.1191.
15. Citarella F., Fedele G., Roem D., et al. The second exon-encoded factor XII region is involved in the interaction of factor XII with factor XI and does not contribute to the binding site for negatively charged surfaces. Blood. 1998; 92(11): 4198–206.
16. Baglia F.A., Jameson B.A., Walsh P.N. Identification and characterization of a binding site for factor XIIa in the Apple 4 domain of coagulation factor XI. J Biol Chem. 1993; 268(6): 3838–44.
17. Schousboe I. Contact activation in human plasma is triggered by zinc ion modulation of factor XII (Hageman factor). Blood Coagul Fibrinolysis. 1993; 4(5): 671–8.
18. Petersen T.E., Thøgersen H.C., Skorstengaard K., et al. Partial primary structure of bovine plasma fibronectin: Three types of internal homology. Proc Natl Acad Sci U S A. 1983; 80(1): 137–41. DOI: 10.1073/pnas.80.1.137.
19. Mahdi F., Madar Z.S., Figueroa C.D., Schmaier A.H. Factor XII interacts with the multiprotein assembly of urokinase plasminogen activator receptor, gC1qR, and cytokeratin 1 on endothelial cell membranes. Blood. 2002; 99(10): 3585–96. DOI: 10.1182/blood.v99.10.3585.
20. Henderson L.M., Figueroa C.D., Müller-Esterl W., Bhoola K.D. Assembly of contact-phase factors on the surface of the human neutrophil membrane. Blood. 1994; 84(2): 474–82.
21. Hasan A.A., Zisman T., Schmaier A.H. Identification of cytokeratin 1 as a binding protein and presentation receptor for kininogens on endothelial cells. Proc Natl Acad Sci U S A. 1998; 95(7): 3615–20. DOI: 10.1073/pnas.95.7.3615.
22. Bradford H.N., Pixley R.A., Colman R.W. Human factor XII binding to the glycoprotein Ib-IX-V complex inhibits thrombin-induced platelet aggregation. J Biol Chem. 2000; 275(30): 22756–63. DOI: 10.1074/jbc.M002591200.
23. Mahdi F., Shariat-Madar Z., Todd R.F., et al. Expression and colocalization of cytokeratin 1 and urokinase plasminogen activator receptor on endothelial cells. Blood. 2001; 97(8): 2342–50. DOI: 10.1182/blood.v97.8.2342.
24. Røjkaer R., Schousboe I. Partial identification of the Zn2+-binding sites in factor XII and its activation derivatives. Eur J Biochem. 1997; 247(2): 491–6. DOI: 10.1111/j.1432-1033.1997.00491.x.
25. Carpenter G., Cohen S. Epidermal growth factor. J Biol Chem. 1990; 265(14): 7709–12.
26. Gordon E.M., Venkatesan N., Salazar R., et al. Factor XII-induced mitogenesis is mediated via a distinct signal transduction pathway that activates a mitogen-activated protein kinase. Proc Natl Acad Sci U S A. 1996; 93(5): 2174–9. DOI: 10.1073/pnas.93.5.2174.
27. Yamada K.M. Cell surface interactions with extracellular materials. Annu Rev Biochem. 1983; 52: 761–99. DOI: 10.1146/annurev.bi.52.070183.003553.
28. Dunn J.T., Silverberg M., Kaplan A.P. The cleavage and formation of activated human Hageman factor by autodigestion and by kallikrein. J Biol Chem. 1982; 257(4): 1779–84.
29. Cochrane C.G., Revak S.D., Wuepper K.D. Activation of Hageman factor in solid and fluid phases. A critical role of kallikrein. J Exp Med. 1973; 138(6): 1564–83. DOI: 10.1084/jem.138.6.1564.
30. Shatzel J.J., DeLoughery E.P., Lorentz C.U., et al. The contact activation system as a potential therapeutic target in patients with COVID-19. Res Pract Thromb Haemost. 2020; 4(4): 500–505. DOI: 10.1002/rth2.12349.
31. Renné T., Schmaier A.H., Nickel K.F., et al. In vivo roles of factor XII. Blood. 2012; 120(22): 4296–303. DOI: 10.1182/blood-2012-07-292094.
32. Ghebrehiwet B., Silverberg M., Kaplan A.P. Activation of the classical pathway of complement by Hageman factor fragment. J Exp Med. 1981; 153(3): 665–76. DOI: 10.1084/jem.153.3.665.
33. Ghebrehiwet B., Randazzo B.P., Dunn J.T., et al. Mechanisms of activation of the classical pathway of complement by Hageman factor fragment. J Clin Invest. 1983; 71(5): 1450–6. DOI: 10.1172/jci110898.
34. Moreno A.S., Valle S.O., Levy S., et al. Coagulation factor XII gene mutation in Brazilian families with hereditary angioedema with normal C1 inhibitor. Int Arch Allergy Immunol. 2015; 166(2): 114–20. DOI: 10.1159/000376547.
35. Deroux A., Boccon-Gibod I., Fain O., et al. Hereditary angioedema with normal C1 inhibitor and factor XII mutation: A series of 57 patients from the French National Center of Reference for Angioedema. Clin Exp Immunol. 2016; 185(3): 332–7. DOI: 10.1111/cei.12820.
36. Colman R.W. Activation of plasminogen by human plasma kallikrein. Biochem Biophys Res Commun. 1969; 35(2): 273–9. DOI: 10.1016/0006-291x(69)90278-2.
37. Goldsmith G.H. Jr., Saito H., Ratnoff O.S. The activation of plasminogen by Hageman factor (Factor XII) and Hageman factor fragments. J Clin Invest. 1978; 62(1): 54–60. DOI: 10.1172/JCI109113.
38. Mandle R. Jr., Kaplan A.P. Hageman factor substrates. Human plasma prekallikrein: Mechanism of activation by Hageman factor and participation in Hageman factor-dependent fibrinolysis. J Biol Chem. 1977; 252(17): 6097–104.
39. Mandle R. Jr., Kaplan A.P. Hageman-factor-dependent fibrinolysis: Generation of fibrinolytic activity by the interaction of human activated factor XI and plasminogen. Blood. 1979; 54(4): 850–62.
40. Жалялов А.С., Баландина А.Н., Купраш А.Д. и др. Современные представления о системе фибринолиза и методах диагностики ее нарушений. Вопросы гематологии/онкологии и иммунопатологии в педиатрии. 2017; 16(1): 69–82. DOI: 10.24287/1726-1708-2017-16-1-69-82.
41. Loza J.P., Gurewich V., Johnstone M., Pannell R. Platelet-bound prekallikrein promotes pro-urokinase-induced clot lysis: A mechanism for targeting the factor XII dependent intrinsic pathway of fibrinolysis. Thromb Haemost. 1994; 71(3): 347–52.
42. Gurewich V., Johnstone M., Loza J.P., Pannell R. Pro-urokinase and prekallikrein are both associated with platelets. Implications for the intrinsic pathway of fibrinolysis and for therapeutic thrombolysis. FEBS Lett. 1993; 318(3): 317–21. DOI: 10.1016/0014-5793(93)80537-5.
43. Кугаевская Е.В., Гуреева Т.А., Тимошенко О.С., Соловьева Н.И. Система активатора плазминогена урокиназного типа в норме и при жизнеугрожающих процессах (обзор). Общая реаниматология. 2018; 14(6): 61–79. DOI: 10.15360/1813-9779-2018-6-61-79.
44. Petersen L.C., Lund L.R., Nielsen L.S., et al. One-chain urokinase-type plasminogen activator from human sarcoma cells is a proenzyme with little or no intrinsic activity. J Biol Chem. 1988; 263(23): 11189–95. DOI: 10.1016/S0021-9258(18)37940-7.
45. Smith D., Gilbert M., Owen W.G. Tissue plasminogen activator release in vivo in response to vasoactive agents. Blood. 1985; 66(4): 835–9.
46. Brown N.J., Nadeau J.H., Vaughan D.E. Selective stimulation of tissue-type plasminogen activator (t-PA) in vivo by infusion of bradykinin. Thromb Haemost. 1997; 77(3): 522–5.
47. Stead N., Kaplan A.P., Rosenberg R.D. Inhibition of activated factor XII by antithrombin-heparin cofactor. J Biol Chem. 1976; 251(21): 6481–8.
48. Silverberg M., Dunn J.T., Garen L., Kaplan A.P. Autoactivation of human Hageman factor. Demonstration utilizing a synthetic substrate. J Biol Chem. 1980; 255(15): 7281–6.
49. Pixley R.A., Schapira M., Colman R.W. Effect of heparin on the inactivation rate of human activated factor XII by antithrombin III. Blood. 1985; 66(1): 198–203.
50. Berrettini M., Schleef R.R., España F., et al. Interaction of type 1 plasminogen activator inhibitor with the enzymes of the contact activation system. J Biol Chem. 1989; 264(20): 11738–43.
51. Schapira M., Ramus M.A., Jallat S., et all. Recombinant alpha 1-antitrypsin Pittsburgh (Met 358----Arg) is a potent inhibitor of plasma kallikrein and activated factor XII fragment. J Clin Invest. 1986;77(2):635–7. doi: 10.1172/JCI112347.
52. Pixley R.A., Schapira M., Colman R.W. The regulation of human factor XIIa by plasma proteinase inhibitors. J Biol Chem. 1985; 260(3): 1723–9.
53. Scott C.F., Carrell R.W., Glaser C.B., et al. Alpha-1-antitrypsin-Pittsburgh. A potent inhibitor of human plasma factor XIa, kallikrein, and factor XIIf. J Clin Invest. 1986; 77(2): 631–4. DOI: 10.1172/JCI112346 .
54. Chien P., Pixley R.A., Stumpo L.G., Modulation of the human monocyte binding site for monomeric immunoglobulin G by activated Hageman factor. J Clin Invest. 1988; 82(5): 1554–9. DOI: 10.1172/JCI113765.
55. Schmeidler-Sapiro K.T, Ratnoff O.D., Gordon E.M. Mitogenic effects of coagulation factor XII and factor XIIa on HepG2 cells. Proc Natl Acad Sci U S A. 1991; 88(10): 4382–5. DOI: 10.1073/pnas.88.10.4382.
56. LaRusch G.A, Mahdi F., Shariat-Madar Z., et al. Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood. 2010; 115(24): 5111–20. DOI: 10.1182/blood-2009-08-236430.
57. Fernando A.N., Fernando L.P., Fukuda Y., Kaplan AP. Assembly, activation, and signaling by kinin-forming proteins on human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2005; 289(1): H251–7. DOI: 10.1152/ajpheart.00206.2004.
58. Rebuck JW. The skin window as a monitor of leukocytic functions in contact activation factor deficiencies in man. Am J Clin Pathol. 1983; 79(4): 405–13. DOI: 10.1093/ajcp/79.4.405.
59. Jablonska E., Markart P., Zakrzewicz D., et al. Transforming growth factor-β1 induces expression of human coagulation factor XII via Smad3 and JNK signaling pathways in human lung fibroblasts. J Biol Chem. 2010; 285(15): 11638–51. DOI: 10.1074/jbc.M109.045963.
60. Göbel K., Pankratz S., Asaridou C.M., et al. Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87-mediated modulation of dendritic cells. Nat Commun. 2016; 7: 11626. DOI: 10.1038/ncomms11626.
61. Hess R., Wujak L., Hesse C., et al. Coagulation factor XII regulates inflammatory responses in human lungs. Thromb Haemost. 2017; 117(10): 1896–1907. DOI: 10.1160/TH16-12-0904.
62. Roche J.A., Roche R. A hypothesized role for dysregulated bradykinin signaling in COVID-19 respiratory complications. FASEB J. 2020; 34(6): 7265–9. DOI: 10.1096/fj.202000967.
63. Shatzel J.J., DeLoughery E.P., Lorentz C.U., et al. The contact activation system as a potential therapeutic target in patients with COVID-19. Res Pract Thromb Haemost. 2020; 4(4): 500–5. DOI: 10.1002/rth2.12349.
64. Wygrecka M., Jablonska E., Henneke I., et al. Coagulation factor XII mediates fibrotic response to lung injury. Pneumologie. 2015; 69: P05. DOI: 10.1055/s-0035-1551907.
65. Wong M., Jaffar J., McMillan L., et al. CSL312, a novel anti-FXII antibody, blocks FXII-induced IL-6production from primary non-diseased and idiopathic pulmonary fibrosis fibroblasts. Am J Respir Crit Care Med. 2020; 201: A6363.
Яковлева Елена Владимировна, кандидат медицинских наук, гематолог, научный сотрудник клинико-диагностического отделения гематологии и нарушений гемостаза
125167, Москва
Зозуля Надежда Ивановна, доктор медицинских наук, заведующая клинико-диагностическим отделением гематологии и нарушений гемостаза
125167, Москва
Яковлева Е.В., Зозуля Н.И. Физиологическая и патологическая роль фактора свертывания крови XII. Гематология и трансфузиология. 2022;67(4):570-578. https://doi.org/10.35754/0234-5730-2022-67-4-570-578
Yakovleva E.V., Zozulya N.I. Physiological and pathological role of factor XII. Russian journal of hematology and transfusiology. 2022;67(4):570-578. (In Russ.) https://doi.org/10.35754/0234-5730-2022-67-4-570-578
![]()
125167, Москва, Новый Зыковский проезд, 4
ФГБУ «НМИЦ гематологии» Минздрава России
тел.: 8-926-816-3887
e-mail: o.levchenko@htjournal.ru






































