

Содержание
Перейти к:
 Е. В. Яковлева,
Н. И. Коняшина,
Л. А. Горгидзе,
В. Л. Сурин,
О. С. Пшеничникова,
О. А. Полеводова,
М. В. Спирин,
Г. М. Галстян,
Н. И. Зозуля
Е. В. Яковлева,
Н. И. Коняшина,
Л. А. Горгидзе,
В. Л. Сурин,
О. С. Пшеничникова,
О. А. Полеводова,
М. В. Спирин,
Г. М. Галстян,
Н. И. Зозуля https://doi.org/10.35754/0234-5730-2019-64-4-489-503
Перейти к:
Введение. Наиболее распространенными наследственными коагулопатиями являются гемофилия и болезнь Виллебранда. Часто под маской этих заболеваний скрываются редкие наследственные коагулопатии, в том числе наследственный дефицит фактора свертывания крови V (FV).
Цель: описание клинических проявлений и выбора тактики лечения у больных с наследственным дефицитом FV.
Основные сведения. Представлен обзор литературы и трех клинических наблюдений за больными с наследственным дефицитом FV. Обсуждаются вопросы дифференциальной диагностики наследственных коагулопатий, поскольку от точного диагноза зависит выбор гемостатической терапии. Больные с наследственным дефицитом FV требуют постоянного наблюдения гематологом с целью контроля спонтанного или индуцированного геморрагического синдрома, проведения гемостатической терапии при оперативных вмешательствах, беременности, родах.
Конфликт интересов: авторы заявляют об отсутствии конфликта интересов.
Финансирование: исследование не имело спонсорской поддержки.
Яковлева Е.В., Коняшина Н.И., Горгидзе Л.А., Сурин В.Л., Пшеничникова О.С., Полеводова О.А., Спирин М.В., Галстян Г.М., Зозуля Н.И. НАСЛЕДСТВЕННЫЙ ДЕФИЦИТ ФАКТОРА СВЕРТЫВАНИЯ КРОВИ V: КЛИНИЧЕСКИЕ НАБЛЮДЕНИЯ. Гематология и трансфузиология. 2019;64(4):489–503. https://doi.org/10.35754/0234-5730-2019-64-4-489-503
Yakovleva E.V., Konyashina N.I., Gorgidze L.A., Surin V.L., Pshenichnikova O.S., Polevodova O.A., Spirin M.V., Galstyan G.M., Zozulya N.I. CONGENITAL FACTOR V DEFICIENCY: CASE REPORTS. Russian journal of hematology and transfusiology. 2019;64(4):489–503. (In Russ.) https://doi.org/10.35754/0234-5730-2019-64-4-489-503
Фактор свертывания крови V (FV) — проакцелерин — известен как лабильный фактор, обладающий прокоагулянтными и антикоагулянтными свойствами. FV является кофактором протромбиназного комплекса, который катализирует превращение протромбина в тромбин. Антикоагулянтные свойства FV обусловлены его участием в инактивации фактора свертывания крови VIII (FVIII) в комплексе с активированным протеином С (activated protein C — APC) и протеином S (protein S — PS) [1]. FV синтезируется в печени в виде пептида, состоящего из 2196 аминокислотных остатков. В плазме циркулирует около 80 % FV, 20 % обнаруживается в α-гранулах тромбоцитов, где он связан с белком-мультимерином. Содержание FV в α-гранулах, возможно, обусловлено синтезом FV мегакариоцитами и/или эндоцитозом из плазмы [1-4]. FV циркулирует в плазме в виде одноцепочечной молекулы массой 330 кDа, период полужизни составляет 12-30 часов [1, 5]. Структурная организация FV сходна с таковой FVIII. В ней выделяют три А-домена, два гомологичных С-домена и один большой центральный В-домен (А1-А2-В-А3-С1-С2). Активируется FV под действием тромбина и, в меньшей степени, — активированного фактора свертывания крови X (FХа). Тромбин активирует FV, расщепляя его по остаткам Arg в положениях 709, 1018 и 1545. Это расщепление освобождает В-домен, и образуется димерная молекула, состоящая из тяжелой цепи массой 105 кDa (А1- А2) и легкой цепи 71-74 кDa (А3-С1-С2), которые взаимодействуют посредством ионов Са2+ и гидрофобных связей (рис. 1) [6].
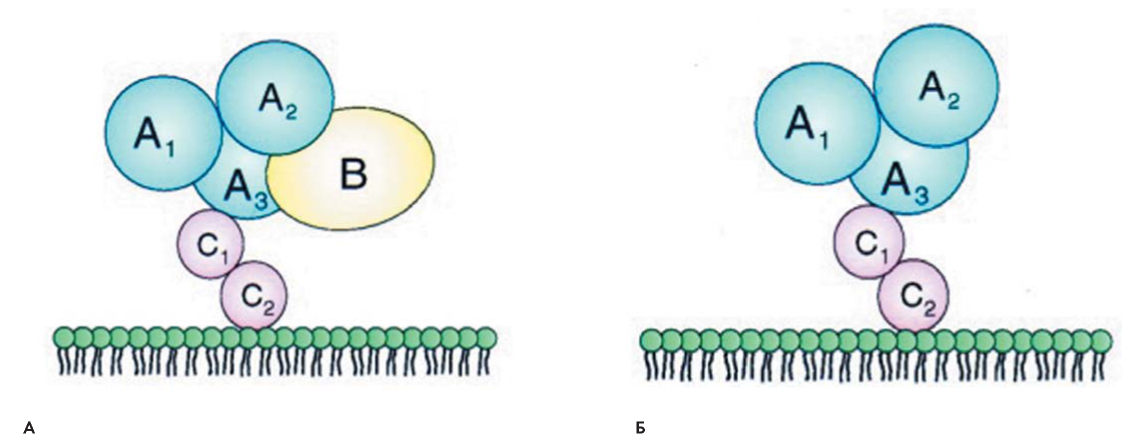
Рисунок 1. Cтруктура FV (А) и FVa (Б) [6]
Figure 1. Structure of FV (А) и FVa (Б)[6]
Активированный FV (FVa) является кофактором протромбиназного комплекса — мультикомпонентного энзимного комплекса, включающего наряду с FVa, FXa, ионы Са2+ и фосфолипиды. Тяжелая цепь FVa обеспечивает контакт с FXa и протромбином, С-домены легкой цепи — взаимодействие с фосфолипидной поверхностью, а А3-домен легкой цепи связывает FXa и фосфолипиды. FVa повышает концентрацию FXa на мембранной поверхности тромбоцитов, активируя рецептор FXa и аллостерически изменяя активные центры FXa, что способствует расщеплению протромбина. Таким образом, FVa выполняет роль кофактора для FXa, многократно увеличивая его способность к активации тромбина (рис. 2) [1, 7, 8].
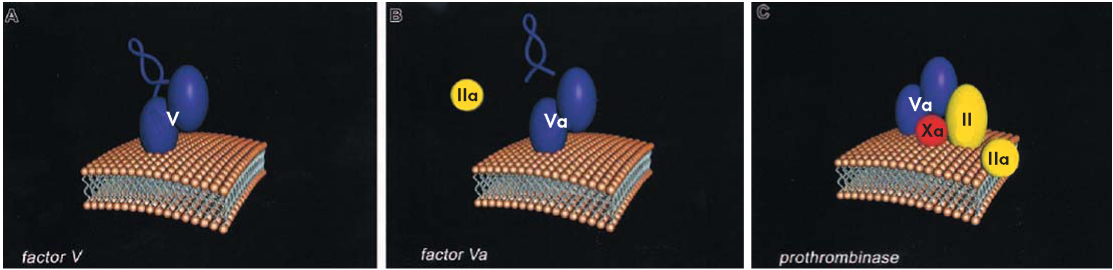
Рисунок 2. Протромбиназный комплекс [8]
Figure 2. Prothrombinase complex [8]
Инактивация FVa осуществляется АРС при его расщеплении по остаткам Arg в положениях 506, 306, 679 тяжелой цепи. Гидролиз по остаткам Arg 306 и Arg 506 полностью инактивирует FV. Гидролиз только по остатку Arg 506 приводит к образованию FV-антикоагулянта (FVac). Тримолекулярный комплекс: АРС, Р8, и FVac элиминирует активность FVIIIa в теназном комплексе посредством его расщепления по остаткам Arg 336, Arg 562, Arg 734 [1, 2, 9]. Способностью инактивировать FVa обладает и α-тромбин, что было показано в исследованиях in vitro и in vivo. Он расщепляет FVa в положении Arg 643, в результате аффинность между легкой и тяжелой цепями значительно снижается [8]. Также эксперименты in vitro демонстрируют способность плазмина инактивировать бычий FVa [10].
Наследственный дефицит FV относится к редким коагулопатиям, распространенность заболевания составляет 1 : 1 000 000 населения. Однако в регионах, где распространены близкородственные браки, частота заболевания возрастает до 1:70 000 населения [11]. Редкие наследственные коагулопатии составляют около 10 % всех наследственных дефицитов факторов свертывания крови. Эпидемиология редких коагулопатий представлена в таблице 1 [12, 13].
Таблица 1. Сравнительная частота наследственного дефицита FV в группе редких наследственных коагулопатий в ряде стран ( %) [12, 13]
Table 1. Comparing the frequency of congenital factor V deficiency with other rare congenital bleeding disorders in some countries ( %j [12, 13]
|
Страна Country |
Дефициты факторов свертывания крови; % больных в группе редких наследственных коагулопатий Blood clotting factor deficiencies, % of patients in the group of rare congenital bleeding disorders |
|||||||
|---|---|---|---|---|---|---|---|---|
|
I |
II |
V |
V+VIII |
VII |
X |
XI |
XIII |
|
|
Италия Italy |
8 |
5 |
10 |
9 |
25 |
8 |
24 |
11 |
|
Иран Iran |
11 |
2 |
7 |
11 |
39 |
10 |
7 |
13 |
|
Индия India |
13 |
3 |
8 |
6 |
15 |
16 |
9 |
30 |
Наследственный дефицит FV был описан норвежским врачом Paul A. Owren во время Второй мировой войны у 29-летней женщины, страдавшей спонтанными экхимозами, профузными носовыми кровотечениями, посттравматическими кровотечениями, меноррагиями и потерей зрения. Изначально это состояние он назвал парагемофилией, определив отсутствие в крови ранее неизвестного прокоагулянта. Впервые эти данные были опубликованы в Норвегии в 1944 г., а после окончания Второй мировой войны — в журнале Lancet в 1947 г. [14]. Заболевание имеет аутосомно-рецессивный путь наследования. В литературе описано более 200 случаев наследственного дефицита FV.
Молекулярные основы заболевания. Полная генетическая структура FV была описана в 1992 г. L.D. Cripe и соавт. [15]. Ген, кодирующий синтез FV, локализуется на хромосоме 1 в локусе 1q24, состоит из 25 экзонов и имеет протяженность 74кb [15, 16]. При дефиците FV идентифицировано более 150 различных мутаций и свыше 700 полиморфизмов, которые не ассоциируются с клиническими проявлениями [1,17]. Среди выявленных мутаций преобладают точечные миссенс- и нонсенс-мутации (65 %), микроделеции (16 %) и мутации сплайсинга (11 %). По данным, приведенным в обзоре R. Asselta и F. Peyvandi [3], из 48 мутаций, вызывающих тяжелую форму заболевания, 21 (44 %) представляли собой микроделеции или микроинсерции, 14 (29 %) — миссенс-мутации, 7 (15 %) — дефекты сплайсинга и 6 (12 %) — нонсенсмутации. Однако вопрос о корреляции между различными мутациями и клиническими проявлениями заболевания остается открытым.
Клинические проявления. Спектр клинических проявлений дефицита FV разнообразен и по источнику кровотечения, и по его интенсивности. Превалирующими являются носовые, десневые, посттравматические, постоперационные кровотечения, гематомы, экхимозы. Гемартрозы — нечастое клиническое проявление дефицита FV, в отличие от наследственного дефицита FVIII. Ж.изнеугрожающими состояниями при дефиците FV являются желудочно-кишечные кровотечения (ЖКК), кровоизлияния в центральную нервную систему (ЦНС).
Согласно Американскому регистру редких наследственных коагулопатий [18], спектр клинических проявлений следующий: 44 % всех эпизодов кровоизлияний — кожный геморрагический синдром, носовые кровотечения и кровоточивость десен, 23 % — кровоизлияния в суставы и мышцы, 19 % — мочеполовые кровотечения, 6 % — ЖКК, 8 % — кровоизлияния в ЦНС. Среди 35 больных иранцев 57 % из них страдали носовыми и десневыми кровотечениями, 50 % женщин имели меноррагии, 29 % имели постоперационные и/или послеродовые кровотечения, 29 % — мышечные гематомы, 26 % — гемартрозы, 6 % — ЖКК; 6 % — кровоизлияния в ЦНС [19].
Корейскими авторами был проведен анализ клинических данных 10 больных. Один из них не имел клинических симптомов заболевания, у 40 % больных отмечены носовые, десневые, постоперационные кровотечения, меноррагии; внутрибрюшное кровотечение констатировано у 2 (20 %) больных, кровоизлияние в ЦНС — у 1 (10 %), ретроперитонеальная гематома — у 1 (10 %) больного и гемартроз также у 1 (10 %) больного. ЖКК, гематурии не зарегистрированы [20]. Индийскими авторами [21] описано 26 больных с наследственным дефицитом FV, из которых 19 (73 %) страдали спонтанным возникновением экхимозов, 5 (19 %) — мышечных гематом, 14 (53 %) — носовых кровотечений, 15 (58 %) — десневых кровотечений. ЖКК имели 10 (38 %) больных, кровотечения из мочеполового тракта — 6 (23 %), кровоизлияния в ЦНС — 6 (23 %) больных. Послеоперационные кровотечения констатированы у 9 (35 %) больных. Подавляющее большинство больных (92 %) отмечали длительные кровотечения после порезов. Все женщины (12 из группы 26 больных) предъявляли жалобы на меноррагии.
Лабораторная диагностика. Характерными лабораторными признаками наследственного дефицита FV являются снижение протромбина по Квику или сочетанные изменения: снижение протромбина по Квику и удлинение активированного частичного тромбопластинового времени (АЧТВ). Диагноз достоверен в случае изолированного снижения активности FV менее 70 % и при отсутствии ингибитора к FV. В зависимости от активности FV в плазме выделяют тяжелую форму дефицита FV (FV менее 1 %), среднюю (FV 1-5 %), легкую (FV более 5 %) [1, 20, 22]. Некоторые авторы диагностируют легкую форму заболевания при активности FV более 10 % [11]. Легкие формы дефицита FV могут сопровождаться изменениями только протромбина по Квику, АЧТВ при этом остается в пределах нормальных значений. В Американском регистре редких наследственных коагулопатий используется классификация гомозиготной и гетерозиготной формы заболевания с пороговым значением FV 20 % [18].
Лечение. Гемостатическая терапия больным с наследственным дефицитом FV назначается с целью лечения геморрагического синдрома (спонтанного или вследствие травмы), периоперационной подготовки, ведения беременности и родоразрешения, регуляции менструальных кровопотерь. Заместительной специфической терапии в настоящее время не существует. Гемостатическая терапия может осуществляться несколькими препаратами. Свежезамороженная плазма (СЗП) является доступным средством, содержащим FV. Дозировка варьирует в пределах 5—20 мл/кг массы тела больного, интервал введения — каждые 12—24 ч в зависимости от интенсивности геморрагического синдрома и индивидуального ответа. Недостатками использования СЗП являются возможности перегрузки объемом, риски инфицирования, появления ингибитора к FV, развития аллергической реакции.
Использование рекомбинантного активированного фактора свертывания VII (эптаког альфа активированного) является применением по неутвержденным показаниям (off-label), и подбор дозы препарата представляет собой определенную трудность. Преимущества использования эптаког альфа (активированного) — это отсутствие риска инфицирования и перегрузки объемом.
Ингибиторы фибринолиза могут быть использованы как в виде пероральной формы, так и внутривенно и позволяют больным проводить гемостатическую терапию в домашних условиях при малоинтенсивном геморрагическом синдроме (при носовых, десневых кровотечениях, меноррагиях). Меноррагии являются основной проблемой у женщин с дефицитом FV и требуют совместного наблюдения с гинекологом. Неэффективность терапии ингибиторами фибринолиза является поводом для решения вопроса о назначении гормональных препаратов.
Описаны случаи эффективного использования концентрата тромбоцитов у больных с дефицитом FV и тяжелыми кровотечениями, которые не отвечали на трансфузии СЗП. Эффективность трансфузий концентрата тромбоцитов и неэффективность СЗП объясняли наличием ингибитора к FV, поскольку содержащийся в тромбоцитах FV не успевал нейтрализоваться ингибитором при высвобождении из активированных тромбоцитов и потреблялся на образование протромбиназного комплекса [1, 3].
Цель настоящей работы — описание клинических проявлений и выбора тактики лечения у больных с наследственным дефицитом FV.
В отделе коагулопатий ФГБУ «НМИЦ гематологии» Минздрава России наблюдаются 170 больных редкими наследственными коагулопатиями. Распределение больных наследственными коагулопатиями по нозологиям представлено в таблице 2. С диагнозом «наследственный дефицит FV» состоят под наблюдением 8 больных (5 женщин, 3 мужчин).
Таблица 2. Распределение больных редкими наследственными коагулопатиями, состоящих на учете в отделе коагулопатий НМИЦ гематологии
Table 2. Distribution of patients with rare inherited bleeding disorders in the Department of coagulopathy disorders of the National Research Center for hematology
|
Дефицит фактора свертывания крови Blood clotting factor deficiency |
Число больных, n ( %) Number of patients, n ( %) |
|---|---|
|
I |
29 (17) |
|
II |
1 (0,5) |
|
V |
8 (5) |
|
V+VIII |
4 (2) |
|
VII |
82 (48) |
|
X |
4 (2) |
|
XI |
25 (15) |
|
XIII |
10 (6) |
|
Комбинированные дефициты Combined coagulation factor deficiencies |
7 (4) |
Больной Л., 30 лет, впервые обратился в НМИЦ гематологии в 2012 г., когда был доставлен бригадой «скорой медицинской помощи» и госпитализирован в связи с коагулопатией неясного генеза и спонтанной напряженной гематомой левого предплечья. Тяжесть состояния была обусловлена обширной наряженной гематомой в области левого предплечья, локализовавшейся на наружной и внутренней поверхностях предплечья от лучезапястного до локтевого суставов. В связи с многократным удлинением АЧТВ и снижением протромбина по Квику были исключены наиболее распространенные наследственные коагулопатии — гемофилия, болезнь Виллебранда и начата гемостатическая терапия СЗП.
Анамнез заболевания. Больной — уроженец Республики Мордовия. Родственный брак родителей отрицает. Семейный анамнез по кровоточивости был не отягощен. В возрасте 1 года получил травму губы, отмечалось выраженное кровотечение, был госпитализирован, с гемостатической целью проводилась электрокоагуляция. В 1993 г., в возрасте 7 лет, был направлен в Измайловскую детскую клиническую городскую больницу (г. Москва), где был установлен диагноз «Болезнь Виллебранда». В клинической картине заболевания отмечал гематомы, экхимозы, дискомфорт в голеностопных суставах. В 1995 г. лечился в реанимационном отделении в связи с ЖКК. В 2008 г. впервые выявлены антитела к вирусу гепатита С. Трансфузионный анамнез: с 1992 г. осуществлялись трансфузии СЗП, криопреципитата, эритроцитной массы по требованию.
При обследовании в НМИЦ гематологии в коагуло- грамме выявлено: АЧТВ 71 с, фибриноген 3,0 г/л, протромбин по Квику 48 %, FV 5 %, FII 98 %, FVII 90 %; FVIII 84 %, FIX 78 %, FX 96 %, FXI 79,9 %, FW 130 %, FXII 101 %, XIIа-зависимый фибринолиз 6 мин, агрегация тромбоцитов с ристомицином 85 %, агрегация тромбоцитов с коллагеном 73 %, агрегация тромбоцитов с адреналином 62 %, агрегация тромбоцитов с АДФ 69 %. Отсутствие ингибитора к FV позволило исключить приобретенный дефицит FV. В общем анализе крови у больного гемоглобин был 132 г/л, эритроциты 4,4 х 1012/л, тромбоциты 174 х 109/л; лейкоциты 6 х 109/л; в биохимическом анализе крови — общий белок 69 г/л, альбумин 40 г/л, аланинаминотрансфераза 42 ед/л, аспартатамино- трансфераза 29 ед/л, креатинин 85 мкмоль/л. Были выявлены антитела к вирусу гепатита С.
Учитывая анамнестические, клинические и лабораторные данные, у больного был диагностирован наследственный дефицит FV. Продолжены трансфузии СЗП в дозе 700 мл/сутки (8 мл/кг массы тела), после стабилизации состояния больного суточная доза СЗП была уменьшена, он был выписан из НМИЦ гематологии спустя 7 суток.
Контрольное обследование больного было проведенное через месяц после окончания гемостатической терапии и позволило установить тяжелую форму заболевания по лабораторным данным: FV 0,4 %, АЧТВ 155 с, протромбин по Квику 12,6 %. По данным тромбоэластографии (ТЭГ), ротационной тромбоэластометрии (РОТЭМ) с оценкой внешнего (EXTEM) и внутреннего (INTEM) путей свертывания крови у больного были выявлены признаки гипокоагуляции (рис. 3). Повторное обращение в НМИЦ гематологии в 2016 г. было вызвано образованием после физического напряжения обширной гематомы на медиальной поверхности бедра справа размерами 10 х 15 см (рис. 4А), а в 2017 г. — в связи с формированием в результате травмы гематомы в области правого локтевого сустава, переходящей на предплечье, размерами 10 х 7 см (рис. 4Б). В 2018 г. поводом для обращения послужили боли и отечность в области левого голеностопного сустава, при обследовании был диагностирован гемартроз. Во всех случаях больному проводилась заместительная гемостатическая терапия СЗП в дозе 7-10 мл/кг с выраженным положительным эфф ектом. В ноябре 2018 г. у больного появились периодические приступообразные боли в поясничной области, купировавшиеся спазмолитиками. По результатам обследования, включавшего ультразвуковое исследование (УЗИ), магнитно-резонансную томографию с внутривенным контрастированием, была диагностирована мочекаменная болезнь, выявлен камень в лоханочно-мочеточниковом сегменте правой почки. Гематурия и очередной приступ почечной колики явились показанием для экстренной госпитализации и решения вопроса о тактике лечения. Рассматривались следующие варианты лечения: дистанционная ударно-волновая литотрипсия, пиелолитотомия, контактная литотрипсия. Учитывая высокий риск геморрагических периоперационных осложнений, при выборе метода лечения учитывали как урологические, так и гематологические факторы, поэтому 16 ноября 2018 г. больному была выполнена контактная лазерная уретеролитотрипсия справа со стентированием правого мочеточника. В периоперационном периоде в качестве гемостатической терапии проводились ежедневные трансфузии СЗП в объеме 600-700 мл/сут. Благодаря такой терапии перед операцией протромбин по Квику составил 47 %, АЧТВ 52 с. Однако после оперативного вмешательства у больного сохранялись болевой синдром, требовавший повторного введения спазмолитиков и анальгетиков, и гематурия, что расценили как следствие нарушения гемостаза, а не урологических причин. Гемостатическая терапия СЗП была продолжена в прежнем объеме, АЧТВ поддерживалось в пределах 50 с, протромбин по Квику — 45 %. Увеличение объема трансфузии СЗП представлялось опасным из-за возможного развития тампонады мочевого пузыря сгустками крови. Однако болевой синдром и макрогематурия сохранялись. В связи с нефункциональным стентом 26.11.2018 выполнена процедура удаления стента правого мочеточника, который оказался частично тромбирован. Гемостатическая терапия была отменена. Состояние больного стабилизировалось, был выписан, приступил к трудовой деятельности.
Срок наблюдения за больным составляет 7 лет.
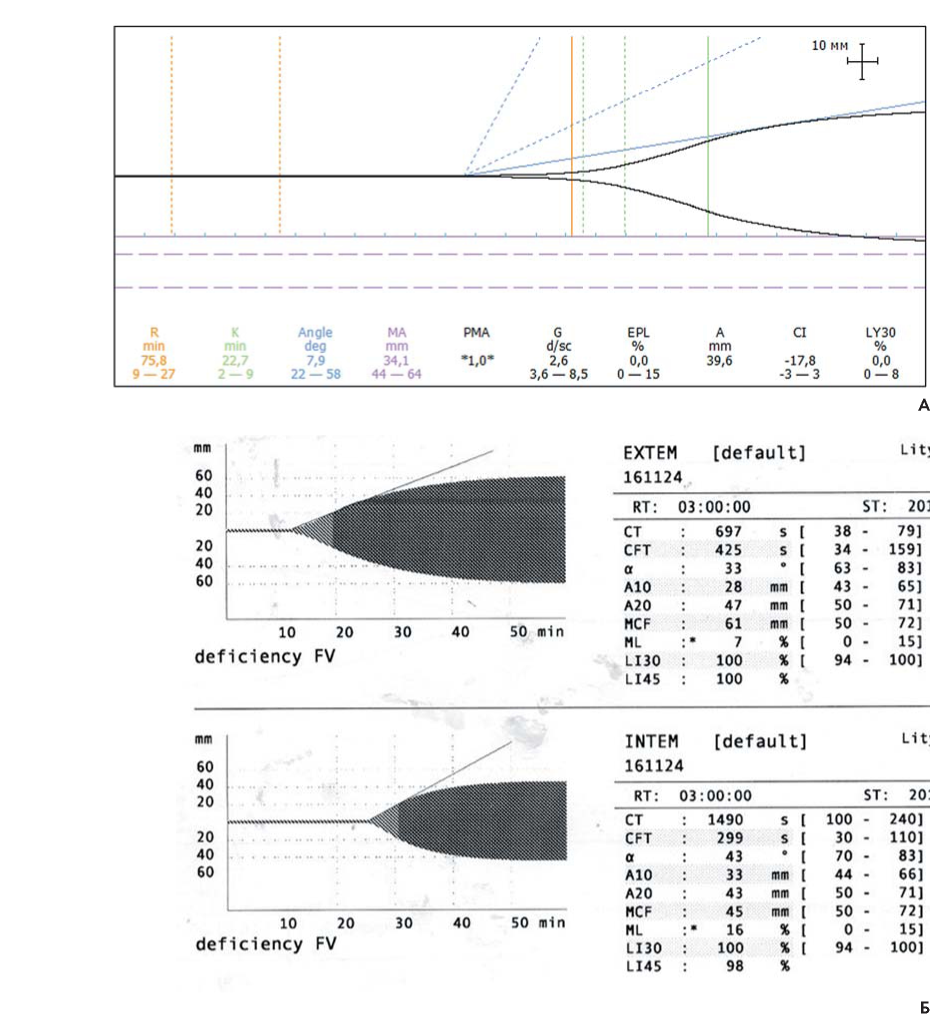
Рисунок 3. Интегральные тесты больного Л. А. Тромбоэластограмма с цельной цитратной кровью. Б. Ротационная тромбоэластометрия (EXTEM, INTEM)
Figure 3. Integral hemostatic tests of patient L. A. Thromboelastogram with whole citrate blood. Б. Rotational thromboelastometry (EXTEM, INTEM)

Рисунок 4. Геморрагический синдром у больного Л. с наследственным дефицитом фактора свертывания крови V. А. Обширная гематома медиальной области бедра справа. Б. Гематома области локтевого сустава с переходом на предплечье
Figure 4. Hemorrhagic syndrome in patient L. with congenital FV deficiency. А. Extensive hematoma of medial area of a hip on the right. Б. Hematoma of the elbow joint with the transition to the forearm
Больная Р. в детском возрасте отмечала спонтанное появление синяков на коже туловища и конечностей, носовые кровотечения. Менструации дебютировали в возрасте 15 лет, они были обильные, продолжительные, менструальный цикл — нерегулярный. В 2011 г., в возрасте 20 лет, она пребывала на стационарном лечении в гинекологическом отделении с диагнозом: «Острый двусторонний сальпингоофорит с нарушением менструальной функции. Эктопия шейки матки. Хронический цервицит. Хронический эндометрит. Кистозно-измененные яичники». Проводилась консервативная терапия с положительным эффектом. При обследовании на тот момент АЧТВ составляло 43 с, фибриноген 2,4 г/л, тромбиновое время (ТВ) 19 с, протромбин по Квику колебался от 16,5 до 58 %. Однако, несмотря на выявленные нарушения гемостаза, больная не была консультирована гематологом.
В 2014 г. проведена криодеструкция эрозии шейки матки, геморрагических осложнений не было. В 20142015 гг. с целью регуляции менструальных кровопотерь по рекомендации гинеколога использовала гормональные интравагинальные препараты, принимала гормональные оральные контрацептивы, однако в дальнейшем отказалась от их приема из-за выраженных побочных эффектов, проявлявшихся головными болями, сменами настроения. Семейный анамнез по кровоточивости не отягощен.
В 2015 г., в возрасте 24 лет, в связи с подозрением на болезнь Виллебранда она была направлена для обследования в отдел коагулопатий НМИЦ гематологии. Основной жалобой на момент обращения были скудные кровянистые выделения из половых путей в течение трех месяцев. При обследовании у больной АЧТВ было 35 с, фибриноген 3,5 г/л, протромбин по Квику 53 %, ТВ 15 с, плазменная активность FII 103 %, FV 23 %, FVII 172 %, FX 107 %, FVIII 122 %, FW 180 %, XIIa-зависимый фибринолиз 8 мин, агрегация тромбоцитов с АДФ 83 %, агрегация тромбоцитов с ристомицином 88 %, агрегация тромбоцитов с коллагеном 85 %, агрегация тромбоцитов с адреналином 77 %. В общем анализе крови концентрация гемоглобина была 115 г/л, эритроцитов 4,4 х 1012/л, тромбоцитов 308 х 109/л, лейкоцитов 8,1 х 109/л. Биохимические показатели крови были в пределах нормальных значений. Таким образом, лабораторные данные позволили исключить диагноз «болезнь Виллебранда» и предположить наличие у больной наследственного дефицита FV.
С целью генетического подтверждения диагноза в лаборатории генной инженерии НМИЦ гематологии выполнен анализ первичной структуры функционально значимых участков гена FV больной Р. Секвенирование выявило наличие гетерозиготной миссенс-мутации в экзоне 13: CD684 TGT-TAT (Cys684Tyr). Согласно Human Gene Mutation Database (HGMD) [17] данная мутация была описана впервые китайскими авторами в 2008 г. [20].
По данным УЗИ малого таза патологии не было выявлено. На основании осмотра гинекологом диагностированы дисфункция яичников, маточное кровотечение.
Таким образом, на основании лабораторных, клинических и анамнестических данных был диагностирован наследственный дефицит FV, легкая форма. Проведена гемостатическая терапия эптаког альфа (активированным) 3,6 мг в/в (60 мкг/ кг массы тела) без побочных реакций. Назначена антифибринолитическая терапия транексамом 500 мг х 3 раза в день, ангиотропная терапия этамзилатом (дициноном) 250 мг х 4 раза в день. В результате удалось скорректировать менструальный цикл.
Через полтора года больная повторно обратилась с жалобами на обильные менструации продолжительностью до 10 дней. Гемостатическую терапию в течение этих полутора лет не проводила. Проведенное обследование не выявило значительных изменений по сравнению с предыдущими результатами. Активность FV в плазме составила 34 %, протромбин по Квику 53 %, АЧТВ 36 с. Концентрация гемоглобина крови повысилась до 125 г/л.
Были выполнены вискоэластичные тесты оценки гемостаза: ТЭГ и РОТЭМ. По данным ТЭГ показатели начального тромбообразования, динамики свертывания крови, плотности тромба и скорости его лизиса были в пределах референсных значений. По данным РОТЭМ тесты внутреннего (INTEM) и внешнего (EXTEM) путей свертывания крови также были в пределах нормальных значений (рис. 5). Больной возобновлена антифибринолитическая и ангиотропная терапия. На этом фоне отмечено уменьшение менструальной кровопотери.
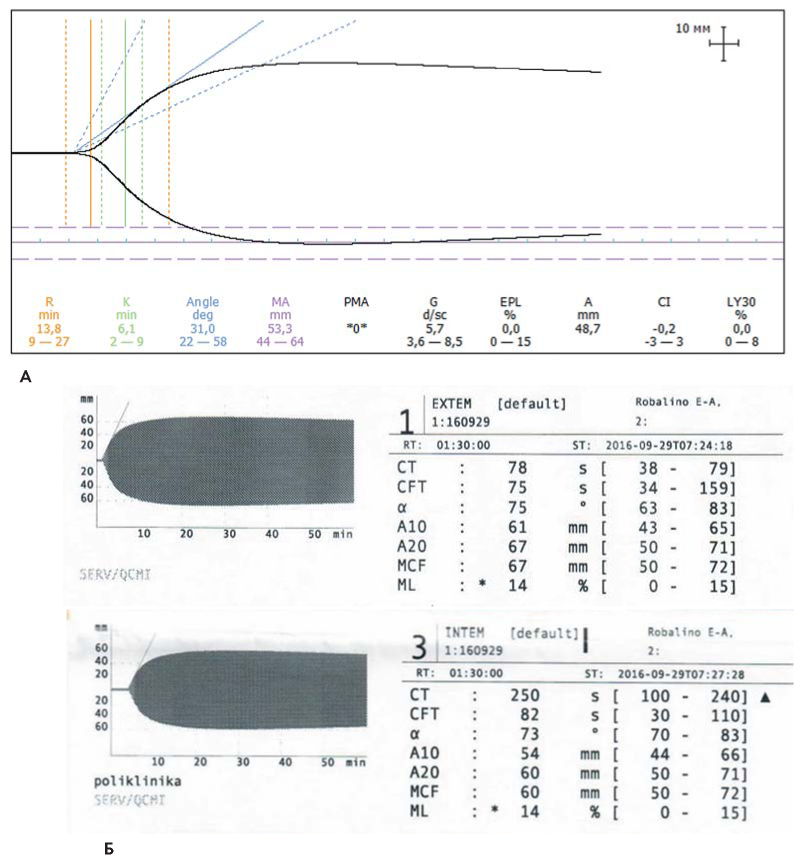
Рисунок 5. Интегральные гемостатические тесты больной Р. А. Нативная тромбоэластограмма с цельной цитратной кровью. Б. Ротационная тромбоэластометрия (EXTEM, INTEM)
Figure 5. Integral hemostatic tests of patient R. A. Thromboelastogram with whole citrate blood. Б. Rotational thromboelastometry (EXTEM, INTEM)
При обращении в январе 2018 г. жалоб не предъявляла, менструации были умеренные, длились 10 дней. По лабораторным данным: АЧТВ 38 с, протромбин по Квику 65 %, плазменная активность FV 41,6 %. В общем анализе крови: гемоглобин 127 г/л, тромбоциты 268 х 109/л, лейкоциты 6,47 х 109/л.
Срок наблюдения за больной составляет 4 года.
Больная И. впервые обратилась в отдел коагулопа- тий НМИЦ гематологии в 2018 г. в возрасте 19 лет с жалобами на нарушения менструального цикла, постоянные кровянистые выделения из половых путей, носовые кровотечения, легкое образование синяков. Впервые наследственный дефицит фактора свертывания крови V был диагностирован у нее в 2005 г. в возрасте 7 лет в г. Санкт-Петербурге. Наблюдалась по месту жительства в г. Череповце.
Анамнез по кровоточивости у больной был осложнен. После рождения наблюдалось длительное заживление пупочной ранки. При прорезывании зубов отмечалась кровоточивость. В возрасте 1 года находилась на лечении в реанимационном отделении в связи с ЖКК, осуществлялись трансфузии эритроцитной массы, аминокапроновой кислоты. В возрасте 3 лет после травмы, которую она получила при катании на качелях, кровотечение из раны продолжалось в течение 3 дней, что наряду с проводимой гемостатической терапией (аминокапроновая кислота, гемостатическая губка) потребовало наложения скоб. В возрасте 4 лет была госпитализирована по поводу эрозивного гастрита. В период с 5 до 10 лет отмечались рецидивирующие гематурии, в связи с чем 12 раз была госпитализирована. В результате геморрагического синдрома у больной развилась постгеморрагическая анемия (концентрация гемоглобина крови 38 г/л). Осуществлялись трансфузии СЗП, эритроцитной массы. В возрасте 17 лет развилась обширная гематома голени и стопы после падения, что также потребовало госпитализации и проведения гемостатической терапии СЗП. Трехкратно выполняли удаление зубов, при этом проводилась гемостатическая терапия СЗП, гемостатической губкой.
Гинекологический анамнез. Менструации — с 11 лет, обильные, цикл — нерегулярный. Многократно пребывала в гинекологических стационарах. В возрасте 12 лет ей была назначена заместительная гормональная терапия эстроген-гестагенными препаратами, которую продолжает по настоящее время. В 2011 г. госпитализировалась в Измайловскую детскую клиническую больницу (г. Москва) в связи с маточным кровотечением, где ей выполняли трансфузии СЗП, вводили эптаког альфа (активированный) с некоторым положительным эффектом. С 2016 г. по настоящее время — многократные госпитализации в связи с маточными кровотечениями, проводилась терапия СЗП, эптакогом альфа (активированным). Сопутствующими проблемами проведения гемостатической терапии являлись реакции на введение СЗП, трудности обеспечения венозного доступа. В 2017 г. на фоне массивной гемотрансфузионной терапии развился флебит левой локтевой вены. В свою очередь, постоянные кровопотери приводили к ане- мизации больной и необходимости курсового проведения терапии препаратами железа.
Семейный анамнез по кровоточивости не отягощен; родственный брак родителей отрицает.
В 2019 г. в связи с регулярной необходимостью проведения гемостатической терапии и отсутствием венозного доступа встал вопрос о необходимости установки порт-системы. С этой целью больная была госпитализирована в НМИЦ гематологии. При обследовании: АЧТВ 176 с, фибриноген 3,8 г/л, протромбин по Квику 11 %, МНО 5,19, ТВ 15,6 с, плазменная активность FII 89,1 %, FV 0,4 %, FVII 110 %, FVIII 183 %, FIX 84, 9 %, FX 119 %, FXI 76,6 %, FXII 114 %, FW 120 %, FXIII 65,7 %, ингибитор к FV не выявлен. В общем анализе крови: концентрация гемоглобина 119 г/л, эритроцитов 4,46 х 1012/л, тромбоцитов 247 х 109/л, лейкоцитов 7,9 х 109/л. Биохимические показатели были в пределах нормальных значений. Выполнены тесты РОТЭМ, в которых гипокоагуляция была обнаружена как в тесте EXTEM (EXTEMСT 484 с, норма 38-79, EXTEMcft 201 с, норма 34-159), так и INTEM (INTEMСT 2290 с, норма 38-79), СFT 519 с, норма 34-159), что объясняется участием FV в свертывании как по внешнему пути, отражающемуся в тесте ЕХТЕМ, так и в свертывании по внутреннему пути, что отражается на тесте INTEM (рис. 6 А).
Учитывая выраженную гипокоагуляцию, перед операцией больной была осуществлена трансфузия 800 мл СЗП и 4 доз концентрата тромбоцитов, после чего был достигнут гемостаз, произведена катетеризация правой внутренней яремной вены и под кожу на грудной клетке имплантирован резервуар порт-системы. В результате гемостатической терапии к началу операции имплантации порт-системы отмечено сокращение АЧТВ до 42 с, увеличение протромбина по Квику до 47 %, плазменной активности FV до 20 %. Показатель EXTEMСT сократился до 116 с, EXTEMСFT до 55 с, INTEMСT до 268 с, INTEMСFT до 78 с (рис. 6Б). На вторые сутки осуществлена повторная трансфузия концентрата тромбоцитов. Гемостатическая терапия СЗП была продолжена в течение 6 дней. Геморрагических осложнений, признаков гематомы в области установки резервуара порт-системы не отмечалось. Больная была выписана домой в г. Череповец.
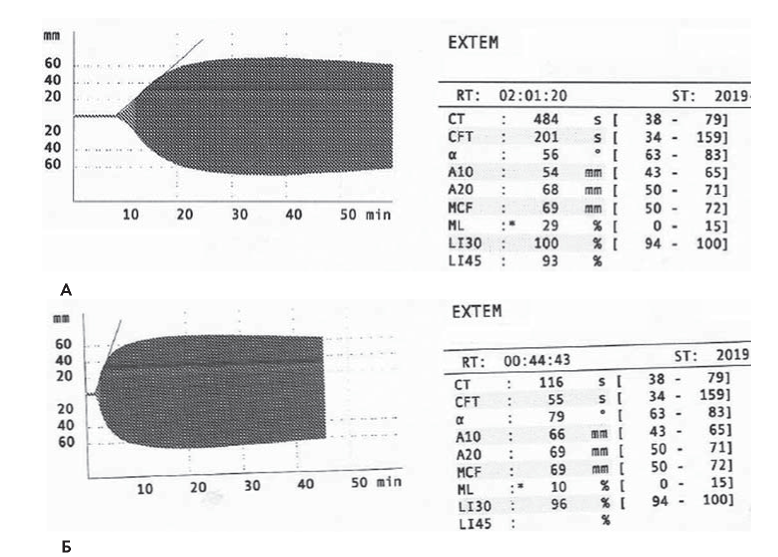
Рисунок 6. Интегральные гемостатические тесты больной И. А. Ротационная тромбоэластометрия (EXTEM) до операции Б. Ротационная тромбоэластометрия (EXTEM) через 30 минут после введения СЗП и концентрата тромбоцитов
Figure 6. Integral hemostatic tests of patient I. A. Rotational thromboelastometry (EXTEMj before surgery. Б. Rotational thromboelastometry (EXTEMj 30 minutes after FFP and platelet concentrate administration
Вопросы диагностики редких наследственных коагулопатий заслуживают особенного внимания. Удлинение АЧТВ и снижение протромбина по Квику могут быть признаками нарушения функции печени, коагулопатии потребления, наследственных дефицитов факторов свертывания крови или наличия ингибиторов к факторам свертывания крови. Дифференциальная диагностика требует расширенного лабораторного исследования параметров внутреннего и внешнего путей свертывания крови.
При заболеваниях печени, сопровождающихся развитием печеночной недостаточности, отмечается уменьшение плазменной концентрации фибриногена, протромбина по Квику, однако плазменная активность FVIII часто повышена.
Коагулопатия потребления — это состояние гиперкоагуляции, развивающееся вследствие различных причин и характеризующееся снижением активности факторов свертывания в плазме крови. Примерами коагулопатий потребления могут быть гестационная коагулопатия, синдром диссеминированного внутри- сосудистого свертывания.
Редкие наследственные коагулопатии характеризуются изолированным снижением активности какого- либо фактора свертывания крови. Степень уменьшения активности фактора свертывания крови может быть различной — от легкой до тяжелой. Это, в свою очередь, по-разному влияет на изменения параметров гемостаза. В первом и третьем клинических наблюдениях плазменная активность FV составляла менее 1 %, протромбин по Квику был снижен до 12 %, АЧТВ составляло более 150 с. Во втором клиническом наблюдении плазменная активность FV составляла 34 %, протромбин по Квику 54 %, а АЧТВ оставалось в пределах нормальных значений. Среди редких наследственных коагулопатий частота сочетанного дефицита факторов V и VIII составляет 3-11 % [12, 13, 24]. Поэтому определение активности FVIII является обязательным для дифференциальной диагностики наследственного дефицита FV. Сочетанный дефицит факторов V и VIII также имеет аутосомно-рецессивный тип наследования. Однако молекулярные основы связаны с наличием мутаций в генах LMANl (Lectin Mannose Binding Protein) или MCFD2 (Multiple Coagulation Factor Deficiency 2), что приводит к дефициту протеинового комплекса, являющегося карго-рецептором транспортировки FV и FVIII от эндоплазматического ретику- лума к комплексу Гольджи [24-26]. Характер клинических проявлений не позволяет идентифицировать эти наследственные коагулопатии, а гемостатическая терапия имеет свои отличия.
Наконец, дифференциальный диагноз следует проводить между наследственным и приобретенным дефицитом FV. В литературе описано около 159 случаев приобретенного дефицита FV [20, 27]. Причины появления ингибитора к FV остаются неизвестными. Обнаружение ингибитора к FV часто ассоциируется с хирургическими процедурами, использованием антибактериальных препаратов, аутоиммунными, онкологическими, инфекционными заболеваниями (туберкулез, ВИЧ). Согласно исследованию Viroj Wiwanitkit [28], из 33 случаев диагностики приобретенного дефицита FV ингибитор к FV был обнаружен у 3 больных с инфекционными заболеваниями, у 3 — с онкологическими заболеваниями, у 2 — с аутоиммунными заболеваниями. У 12 больных предполагается лекарственно-индуцированный генез приобретенного дефицита FV; у 13 — вероятная причина не установлена. Приобретенный дефицит FV чаще диагностируется в зрелом или пожилом возрасте, характеризуется отсутствием семейного геморрагического анамнеза. В отличие от приобретенного дефицита наследственный дефицит FV дебютирует кровотечениями в детском и молодом возрасте, у ряда больных можно выявить семейный характер кровоточивости, родственные браки в генеалогическом древе. Лабораторным дифференциальным диагностическим критерием является отсутствие нормализации АЧТВ в тестах со смешением исследуемой и нормальной донорской плазм (1:1) и измерением остаточной активности FV в единицах Бетезды [20].
Одним из дискуссионных вопросов является корреляция между активностью FV в плазме и интенсивностью геморрагических проявлений. Приведенные в настоящей работе клинические наблюдения демонстрируют соответствие лабораторных данных клиническим проявлениям. Больные в первом и третьем клинических наблюдениях с тяжелой формой FV имели тяжелые клинические проявления с жизнеугрожающими кровотечениями в анамнезе, больная с легкой формой дефицита FV (клиническое наблюдение 2) — легкие клинические проявления. Однако в различных исследованиях показано, что некоторые больные с тяжелой формой дефицита FV (<1 %) могут быть асимптомны или иметь легкой/средней тяжести клинические проявления, в то время как у больных с легкой формой дефицита FV случаются жизнеугрожающие кровотечения [18, 29]. Описан случай, демонстрирующий различные клинические проявления тяжелого дефицита FV у 2 сиблингов. У одного из них заболевание дебютировало в возрасте 3 недель ЖКК, в дальнейшем клинический фенотип характеризовался редким скудным кожно-слизистым геморрагическим синдромом. У его брата заболевание дебютировало с умбиликального кровотечения при рождении, далее в возрасте 3 месяцев стали возникать спонтанные гемартрозы левого коленного сустава, в 4 месяца — субдуральная гематома слева и фронтальная экстрадуральная гематома нетравматического генеза. Больному проводилась профилактическая заместительная гемостатическая терапия СЗП в течение длительного времени [30].
Другим важным клиническим аспектом являются акушерские проблемы. Клиническая картина дефицита FV имеет гендерные отличия, что обусловлено рисками кровопотери во время менструального цикла и родоразрешения у женщин. Причем кровопо- теря во время родов является ведущим акушерским осложнением [31], в то время как взаимосвязь дефицита FV и невынашивания беременности остается дискутабельной. M. Naderi и соавт. [11] опубликовали данные о заболевании дефицитом FV в двух областях Южного Ирана (Sistance and Baluchestan Province) численностью 2 700 000 человек. В этих двух областях дефицитом FV страдают 40 человек — 19 женщин и 21 мужчина, причем 8 из 19 женщин имели беременности, у 6 из которых случались один или более спонтанные аборты [11]. В другом исследовании, в которое было включено 5 больных женщин, страдавших тяжелой и средней тяжести формой заболевания, у 3 акушерский анамнез был отягощен невынашиванием беременности [32]. Принимая во внимание эти факты, необходимо гематологическое сопровождение на этапах планирования, ведения беременности, родоразрешения и в послеродовом периоде. Учитывая молодой возраст больных женщин в описанных клинических случаях, ожидаемыми являются вопросы, связанные с планированием семьи и возможной беременностью.
Таким образом, наиболее распространенными наследственными коагулопатиями являются гемофилия и болезнь Виллебранда. Часто редкие наследственные коагулопатии, в том числе наследственный дефицит FV, клинически проявляются маской болезни Виллебранда. Это требует проведения дифференциальной диагностики на основании расширенных параметров коагулограммы. Интегральные гемостатические пробы не являются чувствительными к легкой форме заболевания. Достоверным лабораторным диагностическим критерием является снижение плазменной активности FV. Генетические исследования не являются рутинными, но могут быть использованы для подтверждения диагноза. Несмотря на соответствие клинических проявлений и тяжести заболевания по лабораторным данным в описанных случаях, степень снижения активности FV не всегда является предиктором клинических проявлений. Больные с наследственным дефицитом FV требуют пожизненного наблюдения гематологом с целью контроля спонтанного или индуцированного травмой геморрагического синдрома, планирования гемостатической терапии при необходимости хирургических вмешательств, ведении беременности и родоразрешении.
1. Huang J.N., Koerper M.A. Factor V deficiency: a concise review. Haemophilia. 2008; 14(6): 1164–9. DOI: 10.1111/j.1365-2516.2008.01785.x
2. Asselta R., Tenchini M.L., Duga S. Inherited defects of coagulation factor V: the hemorrhagic side. J Thromb Haemost. 2006; 4(1): 26–34. DOI: 10.1111/j.1538- 7836.2005.01590.x
3. Asselta R., Peyvandi F. Factor V defi ciency. Semin Thromb Hemost. 2009; 35(4): 382–9. DOI: 10.1055/s-0029-1225760
4. Duckers C., Simioni P., Rosing J., Castoldi E. Advances in understanding the bleeding diathesis in factor V deficiency. Br J Haematol. 2009; 146(1): 17–26. DOI: 10.1111/j.1365-2141.2009.07708.x
5. Totan M., Albayrak D. Intracranial haemorrhage due to factor V defi ciency. Acta Paediatr. 1999; 88(3): 342–3.
6. Nicolaes G.A.F., Dahlbäck B. Factor V and Thrombotic Disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002; 22: 530–8. DOI: 10.1161/01. ATV.0000012665.51263.B7
7. Бочков В.Н., Добровольский А.Б., Кушлинский Н.Е. Под ред. В.А. Ткачука. Клиническая биохимия. 3-е изд., испр. и доп. М.: ГЭОТАР-Медиа, 2008.264 с.
8. Mann K.G., Kalafatis M. Factor V: a combination of Dr Jekyll and Mr Hyde. Blood. 2003; 101(1): 20–30. DOI: 10.1182/blood-2002-01-0290
9. Corral J., Roldán V., Vicente V. Deep Venous Thrombosis Or Pulmonary Embolism And Factor V Leiden: Enigma Or Paradox. Haematologica. 2010; 95(6): 863–6. DOI: 10.3324/haematol.2010.023432
10. Kalafatis M/, Mann K/G. The role of the membrane in the inactivation of factor Va by plasmin. Amino acid region 307–348 of factor V plays a critical role in factor Va cofactor function. J Biol Chem. 2001; 276(21): 18614–23. DOI: 10.1074/jbc.M007134200
11. Naderi M., Tabibian S., Shamsizadeh M., Dorgalaleh A. Miscarriage and recurrent miscarriage in patients with congenital factor V defi ciency: a report of six cases in Iran. Int J Hematol. 2016; 103(6): 673–5. DOI:10.1007/s12185-016-1981-7
12. Naderi M., Tabibian S., Alizadeh S. et al. Congenital factor V deficiency: comparison of the severity of clinical presentations among patients with rare bleeding disorders. Acta Haematol. 2015; 133(2): 148–54. DOI: 10.1159/000363598
13. Shetty S., Shelar T., Mirgal D. et al. Rare coagulation factor deficiencies: a countrywide screening data from India. Haemophilia. 2014; 20(4): 575–81. DOI: 10.1111/hae.12368
14. Owren P.A. Parahaemophilia: haemorrhagic diathesis due to absence of previously unknown clotting factor. Lancet. 1947; 249: 446–8.
15. Cripe L.D., Moore K.D., Kane W.H. Structure of the gene for human coagulation factor V. Biochemistry. 1992; 31(15): 3777–85.
16. Duckers C., Simioni P., Rosing J., Castoldi E. Advances in understanding the bleeding diathesis in factor V deficiency. Br J Haematol. 2009; 146(1): 17–26. DOI: 10.1111/j.1365-2141.2009.07708.x
17. Human Gene Mutation Database (HGMD), www.hgmd.cf.ac.uk
18. Acharya S.S., Coughlin A., Dimichele D.M. North American Rare Bleeding Disorder Study Group. Rare Bleeding Disorder Registry: deficiencies of factors II, V, VII, X, XIII, fibrinogen and dysfibrinogenemias. J Thromb Haemost. 2004; 2(2): 248–56.
19. Lak M., Sharifan R., Peyvandi F., Mannucci PM. Symptoms of inherited factor V deficiency in 35 Iranian patients. Br J Haematol. 1998; 103: 1067–9.
20. Park Y.H., Lim J.H., Yi H.G. et al. Factor V Deficiency in Korean Patients: Clinical and Laboratory Features, Treatment, and Outcome. J Korean Med Sci. 2016; 31(2): 208–13. DOI: 10.3346/jkms.2016.31.2.208
21. Shetty S., Shelar T., Mirgal D. et al. Rare coagulation factor deficiencies: a countrywide screening data from India. Haemophilia. 2014; 20(4): 575–81. DOI: 10.1111/hae.12368
22. Peyvandi F., Di Michele D., Bolton-Maggs P.H. et al. Project on Consensus Defi nitions in Rare Bleeding Disorders of the Factor VIII/Factor IX Scientifi c and Standardisation Committee of the International Society on Thrombosis and Haemostasis. Classification of rare bleeding disorders (RBDs) based on the association between coagulant factor activity and clinical bleeding severity. J Thromb Haemost. 2012; 10(9): 1938–43. DOI: 10.1111/j.1538-7836.2012.04844.x
23. Cao L.J., Wang Z.Y., Su Y.H. et al. Gene analysis of five inherited factor V deficiency cases. Zhonghua Xue Ye Xue Za Zhi. 2008; 29(3): 145–8 (In Chinese).
24. Spreafi co M., Peyvandi F. Combined FV and FVIII deficiency. Haemophilia. 2008; 14(6): 1201–8. DOI: 10.1111/j.1365-2516.2008.01845.x
25. Patel A.J., Liu H.H., Lager R.A. et al. Successful percutaneous coronary intervention in a patient with combined deficiency of FV and FVIII due to novel compound heterozygous mutations in LMAN1. Haemophilia. 2013; 19(4): 607–10. DOI: 10.1111/hae.12128
26. Zhang B. Recent developments in the understanding of the combined deficiency of FV and FVIII. Br J Haematol. 2009; 145(1): 15–23. DOI: 10.1111/j.1365- 2141.2008.07559.x
27. Kunimoto H., Miyakawa Y., Okamoto S. Acquired factor V deficiency and mini literature review. Haemophilia. 2012; 18(3): e86–7. DOI: 10.1111/j.1365- 2516.2011.02650.x
28. Wiwanitkit V. Spectrum of bleeding in acquired factor V inhibitor: a summary of 33 cases. Clin Appl Thromb Hemost. 2006; 12(4): 485–8. DOI: 10.1177/1076029606293438
29. Mumford A.D., Ackroyd S., Alikhan R. et al. Guideline for the diagnosis and management of the rare coagulation disorders: a United Kingdom Haemophilia Centre Doctors’ Organization guideline on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2014; 167(3): 304–26. DOI: 10.1111/bjh.13058
30. Frotscher B., Toussaint-Hacquard M., Fouyssac F. et al. Severe factor V deficiency in two brothers with different clinical presentations. Haemophilia. 2012; 18(5): e383–5. DOI: 10.1111/j.1365-2516.2012.02902.x
31. Girolami A.1., Scandellari R., Lombardi A.M. et al. Pregnancy and oral contraceptives in factor V deficiency: a study of 22 patients (five homozygotes and 17 heterozygotes) and review of the literature. Haemophilia. 2005; 11(1): 26–30.
32. Younesi M.R, Aligoudarzi S.L. Successful delivery in patients with severe congenital factor V deficiency: a study of five homozygous patients. Haemophilia. 2013; 19(5): e318–20. DOI: 10.1111/hae.12210
Яковлева Елена Владимировна, научный сотрудник, кандидат медицинских наук, врач-гематолог отдела коагулопатий
тел.: +7 (495) 612-29-12
Коняшина Надежда Ивановна, кандидат медицинских наук, врач клинической лабораторной диагностики экспресс-лаборатории отделения реанимации и интенсивной терапии
тел.: +7 (910) 477-58-75
Горгидзе Лана Анзоровна, кандидат биологических наук, старший научный сотрудник клинической лабораторной диагностики экспресс-лаборатории отделения реанимации и интенсивной терапии
тел.: +7 (985) 021-70-21
Полеводова Олеся Алексеевна, врач — анестезиолог-реаниматолог отделения реанимации и интенсивной терапии
тел.: +7 (495) 612-48-59
Спирин Михаил Васильевич, кандидат медицинских наук, врач отделения реанимации и интенсивной терапии
тел.: +7 (926) 983-11-39
Галстян Геннадий Мартинович, доктор медицинских наук, заведующий отделением реанимации и интенсивной терапии
тел.: +7 (495) 612-48-59
Яковлева Е.В., Коняшина Н.И., Горгидзе Л.А., Сурин В.Л., Пшеничникова О.С., Полеводова О.А., Спирин М.В., Галстян Г.М., Зозуля Н.И. НАСЛЕДСТВЕННЫЙ ДЕФИЦИТ ФАКТОРА СВЕРТЫВАНИЯ КРОВИ V: КЛИНИЧЕСКИЕ НАБЛЮДЕНИЯ. Гематология и трансфузиология. 2019;64(4):489–503. https://doi.org/10.35754/0234-5730-2019-64-4-489-503
Yakovleva E.V., Konyashina N.I., Gorgidze L.A., Surin V.L., Pshenichnikova O.S., Polevodova O.A., Spirin M.V., Galstyan G.M., Zozulya N.I. CONGENITAL FACTOR V DEFICIENCY: CASE REPORTS. Russian journal of hematology and transfusiology. 2019;64(4):489–503. (In Russ.) https://doi.org/10.35754/0234-5730-2019-64-4-489-503
![]()
125167, Москва, Новый Зыковский проезд, 4
ФГБУ «НМИЦ гематологии» Минздрава России
тел.: 8-926-816-3887
e-mail: o.levchenko@htjournal.ru






































