

Содержание
Перейти к:
 А. О. Абдуллаев,
Е. А. Степанова,
Т. В. Макарик,
Е. Е. Никулина,
С. А. Треглазова,
С. Р. Горячева,
О. А. Шухов,
А. В. Быкова,
Ж. В. Трацевская,
А. Л. Меликян,
А. М. Ковригина,
А. Г. Туркина,
А. Б. Судариков
А. О. Абдуллаев,
Е. А. Степанова,
Т. В. Макарик,
Е. Е. Никулина,
С. А. Треглазова,
С. Р. Горячева,
О. А. Шухов,
А. В. Быкова,
Ж. В. Трацевская,
А. Л. Меликян,
А. М. Ковригина,
А. Г. Туркина,
А. Б. Судариков https://doi.org/10.35754/0234-5730-2020-65-3-263-280
Перейти к:
Введение. Патогенез миелопролиферативных новообразований ассоциирован с химерным геном BCR-ABL1 или одной из «драйверных мутаций» генов JAK2, MPL и CALR (Calreticulin). Однако в классификации Всемирной организации здравоохранения не указаны миелоидные новообразования с более чем одной «драйверной» генетической аномалией.
Цель — поиск мутаций в генах JAK2, MPL и CALR у больных BCR-ABL1-позитивным хроническим миелоидным лейкозом (ХМЛ), а также оценка кинетики уровня найденных мутаций при терапии ингибиторами тирозинкиназ (ИТК). Материалы и методы. В исследование включены препараты мРНК и ДНК клеток крови и костного мозга 567 больных ХМЛ, проходивших периодический мониторинг уровня транскрипта BCR-ABL1 с 2012 по 2019 гг. Уровень транскрипта BCR-ABL1 был определен посредством высокочувствительной количественной полимеразной цепной реакции в реальном времени. Мутации JAK2V617F и MPLW515L/K были выявлены с использованием количественной аллель-специфической полимеразной цепной реакции в реальном времени. Мутации гена CALR были исследованы фрагментным анализом с последующим секвенированием по Сэнгеру.
Результаты. Сочетание мутаций генов BCR-ABL1, JAK2и CALR среди больных ХМЛ, получавших препараты ИТК, составила 1,23 % (7/567). Из них в 0,88 % (5/567) случаев было выявлено сочетание BCR-ABL1 с JAK2V617F и в 0,35 % (2/567) случаев — сочетание BCR-ABL1 с мутациями гена CALR. При терапии препаратами ИТК в 5 из 7 случаев уровень BCR-ABL1 достиг глубокого молекулярного ответа (МО). У 4 из этих больных была прекращена терапия, и у них по настоящее время сохраняется молекулярная ремиссия. В оставшихся 2 случаях не удалось достичь большого МО, несмотря на применение препаратов ИТК второго поколения.
Заключение. Сочетание химерного гена BCR-ABL1 с мутациями генов Jak2 или CALR является редким событием и составило 0,88 и 0,35 % случаев, соответственно. Сочетание BCR-ABL1 с Jak2V617F и мутациями гена CALR не всегда препятствует достижению большого МО.
Абдуллаев А.О., Степанова Е.А., Макарик Т.В., Никулина Е.Е., Треглазова С.А., Горячева С.Р., Шухов О.А., Быкова А.В., Трацевская Ж.В., Меликян А.Л., Ковригина А.М., Туркина А.Г., Судариков А.Б. Частота сочетания и кинетика уровня транскрипта BCR-ABL1 и аллельной нагрузки мутаций JAK2V617F+ и CALR тип-1, -2 у больных хроническим миелолейкозом. Гематология и трансфузиология. 2020;65(3):253-280. https://doi.org/10.35754/0234-5730-2020-65-3-263-280
Abdullaev A.O., Stepanova E.A., Makarik T.V., Nikulina E.Y., Treglazova S.A., Goryacheva S.R., Shukhov O.A., Bykova A.V., Tratsevskaya Z.V., Melikyan A.L., Kovrigina A.M., Turkina A.G., Sudarikov A.B. Frequency of coexistence and kinetics of the BCR-ABL1 transcript level and allele burden of JAK2V617F and CALR Type 1, 2 gene mutations in patients with chronic myeloid leukemia. Russian journal of hematology and transfusiology. 2020;65(3):253-280. (In Russ.) https://doi.org/10.35754/0234-5730-2020-65-3-263-280
Согласно классификации ВОЗ опухолей гемопоэтической и лимфоидной тканей, химерный ген BCR- ABLl является молекулярным маркером хронического миелоидного лейкоза (ХМЛ), а мутации генов JAK2, CALR и MPL — молекулярными маркерами, характерными для истинной полицитемии (ИП), эссенциальной тромбоцитемии (ЭТ) и первичного миелофиброза (ПМФ) [1]. В большинстве случаев у больных миелопролиферативными новообразованиями (МПН) лейкемический клон несет в себе одну из драйверных мутаций. Тем не менее, по данным литературы, сочетание двух драйверных мутаций при МПН не является редким событием. Химерный ген BCR-ABLl может вторично выявляться у части больных с JAK2V617F+ ИП [2][3][4][5][6][7], ЭТ [8][9][10][11][12][13] и ПМФ [14][15] или, наоборот, после успешной элиминации BCR-ABLl+ клона у больных ХМЛ может вторично выявляться JAK2V617F+ и CALR+ ИП, ЭТ [16] и ПМФ. Частота сочетания химерного гена BCR- ABLl и мутации JAK2V617F в различных популяциях больных ХМЛ сильно варьирует. Если J. Jelinek и соавт. [17] из США при исследовании 99 больных ХМЛ не нашли ни одного случая сочетания транскрипта BCR-ABLl и JAK2V617F, то другие авторы из США, увеличив объем выборки до 1570 больных ХМЛ, выявили сочетание клонов у 0,4 % (6/1570) больных [18]. Частота сочетания транскрипта BCR-ABLl и JAK2V617F у больных ХМЛ в исследованиях немецких авторов составила 0,2 % (23/1487) [19], а у польских — 0,7 % [20]. По данным мексиканского исследования, проведенного на небольшой выборке из 142 больных различными видами МПН, частота сочетания двух клонов составила 12,7 % [21]. Наибольшая частота сочетаний транскрипта BCR-ABLl и JAK2V617F отмечена авторами из Пакистана — 26,7 % [22]. Открытые в 2013 г. мутации 9-го экзона гена CALR выявляются в 70—84 % случаев Jak2 и MPL-негативных ЭТ и ПМФ, в 8 % случаев миелодиспластического синдрома и не выявляются при ХМЛ [23][24][25]. Тем не менее за 2014—2019 гг. описаны 12 клинических наблюдений сочетания химерного гена BCR-ABLl и мутации 9-го экзона гена CALR [26][27][28][29][30][31][32][33][34][35][36][37]. Частота такого сочетания описана в польской популяции больных ХМЛ и составляет 0,17 % [17].
Целью данного исследования явилось определение частоты сочетаний и кинетики уровня транскрипта BCR-ABLl и аллельной нагрузки мутаций генов JAK2, MPL и CALR у больных ХМЛ при терапии ингибиторами тирозинкиназ (ИТК).
В исследование было включено 567 больных ХМЛ, получавших терапию препаратами ИТК, которым проводили мониторинг уровня химерного транскрипта BCR-ABLl в ФГБУ «НМИЦ гематологии» Минздрава России с 2012 по 2019 гг. Выделение мРНК и ДНК осуществлялось набором реагентов компании «Интерлабсервис» (Россия) соглас но инструкции производителя. Количественная оценка уровня транскриптов (Mbcr b3a2 и b2a2) BCR-ABLl была проведена с использованием амплификатора Rotor-Gene О (OIAGEN, Германия) и набора реагентов «АмплиСенс® Лейкоз Квант M-bcr-FRT» («Интерлабсервис», Россия). Результаты анализов рассчитаны по международной шкале с учетом фактора конверсии. Поиск мутаций JAK2V617F и MPLw515lik был осуществлен с использованием количественной аллель-специфической (АС) полимеразной цепной реакции в реальном времени. Мутации гена CALR исследованы фрагментным анализом с последующим секвенированием по Сэнгеру. В исследованиях были использованы наборы реагентов компании «Синтол» (Россия).
В результате исследования образцов мРНК и ДНК клеток крови 567 больных с BCR-ABLl+ ХМЛ мутация JAK2V617F была выявлена у 5 больных, а мутации 9-го экзона гена CALR — у 2 больных. Сочетание BCR-ABLl с мутацией MPLW515L/K не было выявлено ни у одного больного ХМЛ (табл. 1).
Таблица 1. Общие сведения о больных XMJI с мутациями JAK2v:i1 F и CALR тип-1, -2
Table 1. General information about CML patients with JAK2yo] F and CALR type-1, 2 mutations

Примечание. ХМЛ — хронический миелолейкоз, ПМФ — первичный миелофиброз, ЭТ — эссенциальная тромбоцитемия, IS — международная шкала, ПЦО — полный цитогенетический ответ, БМО — большой молекулярный ответ; * — временной интервал (мес) между выявлением двух мутаций.
Note. CML — chronic myeloid leukemia, PMF — primary myelofibrosis, PV — Polycythemia Vera, TKI - tyrosine kinase inhibitor; ET — essential thrombocythemia, IS — international scale, CCyR — complete cytogenetic response, MMR — major molecular response; * — time interval (months) between detection of two mutations.
Клиническое наблюдение 1. Больной Ш. Д.А., 59 лет. У больного в октябре 2012 г. в клиническом анализе крови были выявлены лейкоцитоз (144 х 109/л) и тромбоцитоз (904 х 109/л), в миелограмме — бластные клетки 4 %, расширение гранулоцитарного ростка, базофилы 10,5 %, эозинофилы 8 %. Отмечены гепа- томегалия и спленомегалия (+3 и +15 см из-под края реберной дуги, соответственно). При гистологическом исследовании трепанобиоптата костного мозга обнаружена морфологическая картина ХМЛ и ПМФ (рис. 1 А и В). Молекулярно-генетическое исследование выявило уровень транскрипта BCR-ABLl — 80 % и аллельную нагрузку мутации JAK2V617F — 73 %. Установлен клинический диагноз «ХМЛ, хроническая фаза, промежуточная группа риска по Sokal», и начата терапия иматинибом в дозе 400 мг/сут. Через 8 месяцев после начала терапии иматинибом уровень транскрипта BCR-ABLl снизился в 1000 раз (с 80 до 0,08 %), а аллельная нагрузка мутации JAK2V617F снизилась в 3,5 раза (с 73 до 21 %). При повторном исследовании в ноябре 2013 г. уровень транскрипта BCR-ABLl составил 0,014 %, а мутация JAK2V617F снизилась до 5 %. Учитывая сохраняющиеся тромбоцитоз и спленоме- галию на фоне глубокого молекулярного ответа (МО) по уровню транскрипта BCR-ABLl, а также биклональный характер миелопролиферативного заболевания, с марта 2015 г. к терапии добавлен интерферон альфа в дозе 3 млн МЕ через день. С июня 2018 г. больному прекращена терапия иматинибом и продолжена монотерапия интерфероном альфа в дозе 3 млн МЕ через день. К августу 2019 г. уровень транскрипта BCR- ABLl достиг 0,061 %, т.е. глубокой молекулярной ремиссии, а аллельная нагрузка JAK2V611F стабилизировалась в пределах 28—34 % (рис. 1 С).

Рисунок 1. А — гиперклеточный костный мозг с трехростковой гиперплазией: эритроидный росток с выраженными признаками омоложения, в виде «рассыпающихся» кластеров эритрокариоцитов, присутствовали мегалобластоидные формы; гранулоцитарный росток — на всех этапах дифференцировки. Мегакариоциты полиморфны по размерам и морфологии, располагались разрозненно и в виде единичных рыхлых кластеров. Окраска: гематоксилин и эозин. Ув. *200. В —степень ретикулинового фиброза стромы MF-0 с участками MF-1 менее 30 %. Окраска по Гомори. Ув. *200. С — кинетика уровня транскрипта BCR-ABLl и аллельной нагрузки JAK2V617F
Figure 1. A — hypercellular bone marrow with panmyelosis. Loose clusters of eryfhrokaryocyfes with an increased count of erythroid precursors with the presence of megaloblastoid forms. For granulocytic linage, every stage of differentiation is detectable. Megakaryocytes with notable pleomorphism in size and nuclear morphology tending to form occasional loose clusters. Stain: H&E. Magnification: *200. B — stromal reticulin fibrosis grade MF-0, with less than 30 %. Gomori stain. Magnification: *200. C — kinetics of the transcript level BCR-ABLl and allelic load JAK2V617F
Клиническое наблюдение 2. Больная Б. Е.Н., 61 год. В 2007 г. ей был установлен диагноз ИП на основании эритроцитоза (7,42 х 1012/л), лейкоцитоза (14,9 х 109/л), спленомегалии и морфологического исследования тре- панобиоптата костного мозга (рис. 2 А и В). При молекулярно-генетическом анализе обнаружена мутация JAK2V611F. Больной была начата терапия гидроксикар- бамидом в дозе 1 г 2 раза в день. Тем не менее через 5 лет от начала цитостатической терапии, в марте 2012 г., в крови отмечено нарастание лейкоцитоза до 30,5 х 109/л. В трепанобиоптате костного мозга отмечалось расширение гранулоцитарного ростка за счет промежуточных форм, клеток с незрелой морфологией, более характерной для ХМЛ (рис. 2 А и В). При молекулярно-генетическом исследовании обнаружены транскрипт BCR-ABLl в количестве 49,53 % и мутация JAK2V611F с аллельной нагрузкой 95 %. Клинический диагноз ИП был дополнен ХМЛ, и начата терапия иматинибом в дозе 400 мг/сут. Спустя два с половиной месяца терапии из-за токсического э фф екта иматиниб был заменен на нилотиниб в дозе 800 мг/сут. К марту 2014 г. уровень транскрипта BCR-ABL1 снизился до 0,29 %, а аллельная нагрузка JAK2V611F увеличилась до 100 % (рис. 2 С).

Рисунок 2. А — гиперклеточный костный мозг с трехростковой гиперплазией: эритроидный росток расширен, с выраженными признаками омоложения. Гранулоцитарный росток умеренно расширен, на всех этапах дифференцировки. Мегакариоциты полиморфны по размерам и морфологии, располагаются разрозненно и виде единичных рыхлых кластеров межтрабекулярно. Окраска: гематоксилин и эозин. Ув. *200. В — степень ретикулинового фиброза стромы MF-1. Окраска по Гомори. Ув. *200. С — кинетика уровней транскрипта BCR-ABL1 и аллельной нагрузки JAK2V617F у больной Б. Е.Н.
Figure 2. A — hypercellular bone marrow with panmyelosis. Erythropoiesis is normoblastic with multiple erythroid precursors. Granulopoiesis is moderately expanded, with cells being at various differentiation stages. Numerous megakaryocytes with a significant difference in size and morphology tending to form occasional intertrabecular loose clusters. Stain: H&E. Magnification: *200. B — stromal reticulin fibrosis grade MF-1. Stain: Gomori silver. Magnification: *200. C — kinetics of the BCR-ABLl transcript level and allele burden of JAK2V617F
Клиническое наблюдение 3. Больная Х. Е.С., 51 год. Диагноз ХМЛ у нее был установлен в июне 2001 г. на основании лейкоцитоза (44 х 109/л) и обнаружения при стандартном цитогенетическом исследовании (СЦИ) транслокации t(9;22) (q34;q11) в 100 % метафазных ядер клеток крови. Больной была начата терапия интерфероном альфа в дозе 10 млн МЕ/сут в сочетании с малыми дозами цитарабина. С июня 2003 г. назначен иматиниб в дозе 400 мг/сут, затем в связи с отсутствием цитогенетического ответа — 600 мг/сут. В связи с цитогенетической резистентностью через 4 года лечения иматинибом больная была переведена на терапию ИТК 2-го поколения дазатинибом. Однако за 3 года терапии дазатинибом в максимальной дозе 140 мг/сут удалось получить лишь кратковременный цитогенетический ответ (Ph+ в 66 % метафазных ядер), в связи с чем вновь произведена смена ИТК, и с марта 2010 г. начата терапия нилотинибом в дозе 800 мг/сут. Однако за 4 года терапии нилотинибом не удалось получить полный гематологический ответ, в крови сохранялся умеренный тромбоцитоз (600 х 109/л) и эритроцитоз (5,4 х 1012/л). Удалось получить только частичный цитогенетический ответ (Ph+ в 23 % метафаз) и снижение уровня транскрипта BCR-ABL до 3,36 %. Эти обстоятельства послужили причиной поиска других драй- верных мутаций МПН, и в октябре 2014 г. в клетках крови больной была обнаружена мутация JAK2V611F с аллельной нагрузкой 9,1 %. Ретроспективное исследование аллельной нагрузки мутации JAK2V611F показало, что впервые мутация JAK2V611F с уровнем 1 % появилась в январе 2012 г. и имела тенденцию к неуклонному росту аллельной нагрузки. Гистологическое исследование трепанобиоптата костного мозга выявило картину, характерную для ЭТ (рис. 3 А и В). Таким образом, у больной с BCR-ABL1+ ХМЛ было констатировано наличие второго МПН — ЭТ с мутацией JAK2V611F. Учитывая плохую переносимость терапии интерфероном в прошлом, было рекомендовано продолжить терапию нилотинибом (800 мг/сут) в сочетании с гидрокси- карбамидом (0,5—1 г/сут). В результате проводимого лечения достигнута гематологическая ремиссия, однако за 16 лет терапии препаратами ИТК ни разу не удалось достичь большого МО (БМО). К сентябрю 2019 г. уровень транскрипта BCR-ABL1 составил 3,02 %, а аллельная нагрузка мутации JAK2V617F осталась без изменений и составила 20 % (рис. 3 С).

Рисунок 3. А — нормоклеточный костный мозг: гранулоцитарный росток в умеренном количестве, с преобладанием зрелых форм. Эритроидный росток в достаточном количестве, в виде мелких скоплений эритрокариоцитов нормобластического ряда. Мегакариоциты — в увеличенном количестве, обычных и крупных размеров, с атипичной морфологией, гиперлобулярными нормохромными ядрами, располагаются разрозненно. Окраска: гематоксилин и эозин. Ув. *200. В — степень ретикулинового фиброза MF-0 на большем протяжении. Окраска по Гомори. Ув *200. С — кинетика уровней транскрипта BCR-ABLl и аллельной нагрузки JAK2V617F у больного Х. Е.С.
Figure 3. A — normocellular bone marrow. The granulocytes count lies within the normal range with the predominance of mature forms. Erythroid lineage is sufficient, erythrokaryocytes form small clusters. The megakaryocytes count is elevated; there are normal to large forms, with atypical morphology, hyperlobular normochromic nuclei. No clusters of megacaryocytes. Stain: H&E. Magnification: *200. B — stromal reticulin fibrosis grade MF-0. Stain: Gomori silver. Magnification: *200. C — kinetics of the BCR-ABLl transcript level and allele burden of JAK2V6l7F
Клиническое наблюдение 4. Больная К. Н.Н., 60 лет. В 2002 г. впервые у больной была выявлена спленомегалия (+1 см из-под края реберной дуги), повышенная кровоточивость из мелких ран, чувство тяжести и боль в левом подреберье. Показатели гемограммы были удовлетворительными (гемоглобин — 124 г/л, лейкоциты — 2,9 х 109/л, тромбоциты — 170 х 109/л), и поэтому больной было назначено симптоматическое лечение.
Однако в июне 2004 г. отмечено нарастание лейкоцитоза до 70 х 109/л и спленомегалии, а при гистологическом исследовании трепанобиоптата костного мозга обнаружены морфологические признаки, характерные для ПМФ. Последующие 3 года больная с кратковременным терапевтическим эффектом получала цитостатическую (гидроксикарбамид 1 г/сут) и иммуномодулирующую (интерферон альфа 3 млн МЕ/сут) терапию. Тем не менее в феврале 2007 г. отмечено нарастание лейкоцитоза до 37 х 109/л и спленомегалии (+5 см из- под края реберной дуги), а при цитогенетическом исследовании обнаружена транслокация t(9;22) (q34;q11). Больной установлен диагноз ХМЛ, и с апреля 2007 г.
начата терапия иматинибом в дозе 600 мг/сут. Спустя 3 месяца из-за аллергической реакции (крапивница) был отменен иматиниб, и с августа 2007 г. начата терапия нилотинибом в дозе 800 мг/сут. Через 6 месяцев был достигнут полный цитогенетический ответ (ПЦО), а через год — БМО. Однако, несмотря на достижение ПЦО и БМО, сохранялись выраженная спленомегалия, умеренная анемия и тромбоцитопения, единичные миелоциты и нормобласты в крови, постоянный умеренный внутриклеточный гемолиз. По данным повторного гистологического исследования трепанобиоптата костного мозга, выполненного в июне 2012 г., выявлены изменения, характерные для фиброзной стадии ПМФ (рис. 4 А и В), а молекулярно-генетический анализ показал наличие мутации JAK2V617F с аллельной нагрузкой 59 %. С учетом сохраняющегося стабильного глубокого МО в течение 4 лет было решено отменить нило- тиниб и продолжить терапию только гидроксикарбамидом в дозе 0,5—1 г/сут. Однако в связи нарастанием спленомегалии, портальной гипертензии и явлений гиперспленизма (тромбоцитопения, анемия) в апреле 2013 г. была выполнена спленэктомия. В августе 2019 г. при молекулярно-генетическом анализе транскрипт BCR-ABL1 не обнаружен, а аллельная нагрузка мутации JAK2V617F составила 48 % (рис. 4 С).

Рисунок 4. А — гиперклеточный костный мозг за счет расширения гранулоцитарного и мегакариоцитарного ростков; клеточные элементы расположены в виде «цугов». Гранулоцитарный росток представлен преимущественно зрелыми формами с очаговым увеличением эозинофильных гранулоцитов. Мегакариоциты полиморфны по размерам, с атипичной морфологией, расположены разрозненно и в виде рыхлых и плотных кластеров. Эритроидный росток — в умеренном количестве, представлен кластерами эритрокариоцитов нормобластического ряда. Остеосклероз grade 2-3. Окраска: гематоксилин и эозин. Ув. *200. В — степень ретикулинового фиброза стромы MF-3. Окраска по Гомори. Ув. *200. С — кинетика уровней транскрипта BCR-ABLl и аллельной нагрузки JAK2V6l7Fу больного К. Н.Н.
Figure 4. A — bone marrow is hypercellular due to the expansion of granulo- and megakaryopoesis. Hematopoetic cells are arranged in cords due to dense stomal fibrosis. Granulocytes are predominantly represented by mature forms with a focal increase in eosinophilic granulocytes count. Megakaryocytes vary in size with atypical morphology, noticeably tending to form loose and dense clusters. Erythroid cell count lies within the normal range and the lineage is represented by normoblastic erythrokaryocytes clusters. Osteosclerosis grade 2-3. Stain: H&E. Magnification: *200. B — stromal reticulin fibrosis grade MF-3. Stain: Gomori silver. Magnification: *200. C — kinetics of the BCR-ABLl transcript level and allele burden of JAK2V6l7F
Клиническое наблюдение 5. Больной Д. Л.А., 55 лет. Диагноз ИП установлен в феврале 2011 г. на основание гепатоспленомегалии (печень и селезенка выступали на 1 см из-под края реберной дуги), плеторического синдрома и изменений в анализе крови (гемоглобин — 201 г/л, эритроциты — 9,53 х 1012/л, лейкоциты — 11,65 х 109/л и тромбоциты — 476 х 109/л, СОЭ — 0,5 мм/ч). Больному были рекомендованы эритроцитаферез и циторедуктивная терапия гидроксикарбамидом в дозе 1 г/сут. Однако, несмотря на проводимую терапию, через 3 месяца отмечено нарастание лейкоцитоза с 11,65 х 109 до 50 х 109/л. При СЦИ в 22 % метафазных ядер обнаружена мутация t(9;22) (q34;q11), а молекулярно-генетический анализ выявил транскрипт BCR-ABL1. На основании результатов вышеуказанных исследований установлен клинический диагноз — ХМЛ, хроническая фаза, и начата терапия иматинибом в дозе 400 мг/сут. Спустя 6 месяцев после начала терапии иматинибом был достигнут ПЦО, но сохранялись выраженный плеторический синдром: в крови эритроцитоз (7,2 х 1012/л), тромбоцитоз (701 х 109/л) и лейкоцитоз (11,47 х 109/л) без сдвига лейкоцитарной формулы. В сентябре 2012 г. при гистологическом исследовании трепанобиоптата костного мозга обнаружена выраженная картина ИП (рис. 5 А и В), а молекулярно-генетический анализ показал наличие мутации JAK2V617F с аллельной нагрузкой 70,6 %. Было принято решение увеличить дозу иматиниба с 400 до 600 мг/сут. К сентябрю 2013 г. был достигнут БМО, а аллельная нагрузка на мутацию JAK2V617F снизилась до 56 %. Однако через 4 года после достижения стабильного БМО в мае 2017 г. из-за развития негематологической токсичности (задержка жидкости, нестабильное АД) иматиниб был заменен на интерферон альфа в дозе 3 млн МЕ через день.

Рисунок 5. А — гиперклеточный костный мозг за счет трехростковой гиперплазии: эритроидный росток был представлен в виде «рассыпающихся» скоплений эритрока- риоцитов нормобластического ряда, с признаками омоложения; гранулоцитарный росток на всех этапах дифференцировки. Мегакариоциты — полиморфны по размерам и морфологии, располагались преимущественно разрозненно. Окраска: гематоксилин и эозин. Ув. *200. В — степень ретикулинового фиброза стромы MF-0 с участками MF-1 менее 30 %. Окраска по Гомори. Ув. *200. С — кинетика уровня транскрипта BCR-ABLl и аллельной нагрузки мутации JAK2V6l7Fу больного Д. Л.А.
Figure 5. A — hypercellular bone marrow with panmyelosis. Erythropoiesis is normoblastic, represented by large loose groups of erythroid cells with the "left shift". Granulocytes at every differentiation stage are detectable. Numerous megakaryocytes vary in size and morphology; they are predominantly loosely scattered. Stain: H&E. Magnification: x.B — stromal reticulin fibrosis grade MF-0, with less than 30 % of MF-l areas. Stain: Gomori silver. Magnification: x200. C — kinetics of the BCR-ABLl transcript level and allele burden of JAKCv6m
Клиническое наблюдение 6. Больной К. Ю.А., 66 лет. Клинический диагноз ХМЛ, хроническая фаза был установлен ему в августе 2015 г. на основании лейкоцитоза (17,2 х 109/л), гепатоспленомегалии (печень и селезенка на 2 см выступали из-под реберной дуги) и наличия мутации t(9;22) (q34;q11) в 55 % метафазных ядрах клеток крови. При молекулярно-генетическом анализе уровень химерного транскрипта BCR-ABL1 составил 78,15 %. Больному была начата терапия иматинибом в дозе 400 мг/сут. За 4 месяца терапии иматинибом отмечено снижение количества лейкоцитов до 6,8 х 109/л и уровня транскрипта BCR-ABL1 до 2,79 %, что сопровождалось нарастанием тромбоцитоза с 334 х 109 до 1279 х 109/л. При гистологическом исследовании трепанобиоптата костного мозга были выявлены морфологические признаки ПМФ (рис. 6 А и В), а молекулярно-генетическое исследование обнаружило мутацию CALR c.1099_1150del. p. L367fs*46. К терапии с иматинибом в дозе 400 мг/сут добавлен гидроксикарбамид 1 г/сут. По данным молекулярно-генетических исследований, с января 2017 г. сохраняется БМО, а уровень аллельной нагрузки мутации CALRpl5677j46 остается в пределах 40—50 % (рис. 6 С).
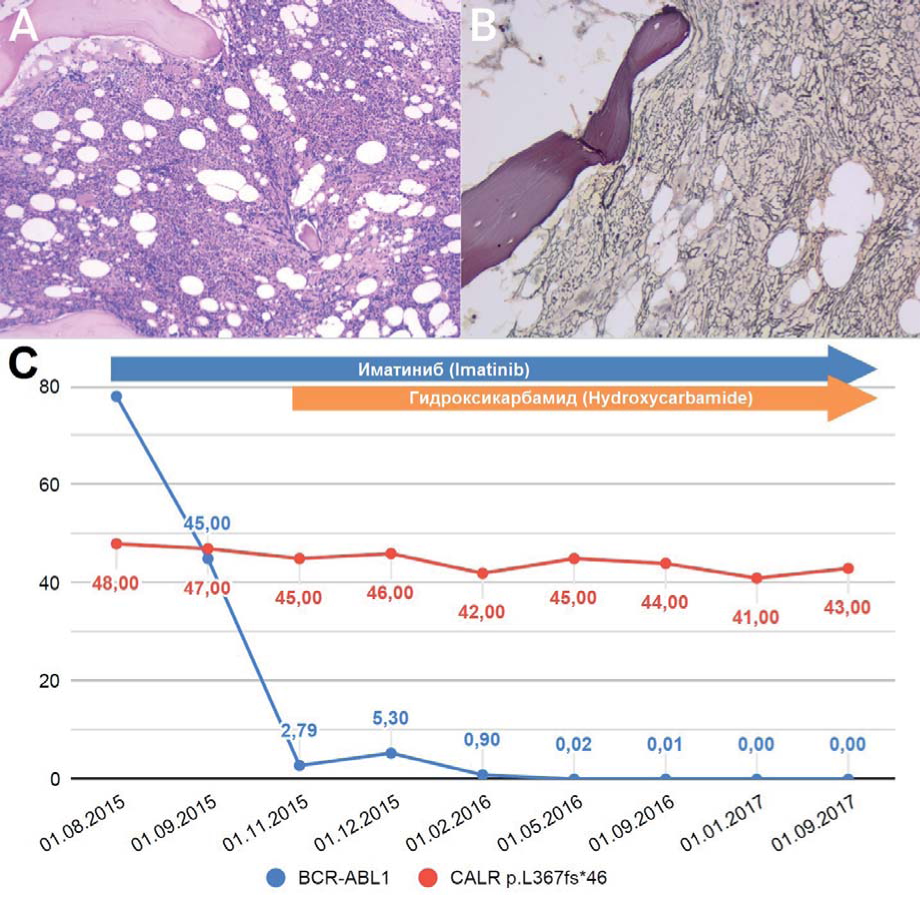
Рисунок 6. А — гиперклеточный костный мозг с гиперплазией гранулоцитарного и мегакариоцитарного ростков. Среди клеток гранулоцитарного ряда преобладают зрелые формы, много клеток эозинофильного ряда. Эритроидный росток в достаточном количестве, часть клеток с омоложением. Отмечается пролиферация мегакариоцитов. Мегакариоциты полиморфны (от крупных клеток со зрелой морфологией, гиперсегментированными ядрами до небольшого размера клеток с гиполобулярными ядрами, нарушенным ядерно-цитоплазматическим соотношением), формируют отдельные рыхлые кластеры. В — при окраске по Гомори степень ретикулинового фиброза MF-2. С — кинетика уровней транскрипта BCR-ABLl и аллельной нагрузки мутации CALRp. L367fs*46
Figure 6. A — hypercellular bone marrow with the expansion of predominantly two cell lineages of myelopoesis. Granulocytic expansion with predominantly mature forms, focal increase of eosinophilic granulocytes count. Erythroid cell count lies within the normal range and the lineage is represented by normoblastic erythrokaryocytes clusters with the admixture of erythroid precursors. Megakaryocytes are polymorphic (from large mature cells with hypersegmented nuclei to small ones with hypolobulated nuclei and disturbance of the nuclear to cytoplasmic ratio), form distinct loose clusters. Stain: H&E. Magnification: x20G.B — stromal reticulin fibrosis grade MF-2. Stain: Gomori silver. Magnification: *200. C — kinetics of the BCR-ABLl transcript level and allele burden of CALRp. L367fs*46
Клиническое наблюдение 7. Больной В. В.С., 66 лет. Диагноз ХМЛ установлен в октябре 2000 г., и до июня 2004 г. больной получал терапию препаратами интерферона альфа. С июля 2004 г. начато лечение иматинибом (400 мг/сут), и через год достигнут БМО. Несмотря на стабильно сохраняющийся БМО, в марте 2017 г. отмечен тромбоцитоз (более 1000 х 109/л). Последующий молекулярно-генетический анализ обнаружил мутацию CALR c.1154_1155insTTGTC (p.K385fs*47) с аллельной нагрузкой 48 %, а гистологическое исследование трепанобиоптата костного мозга выявило морфологическую картину ЭТ. С учетом стабильно сохраняющегося глубокого МО иматиниб был заменен на интерферон альфа в дозе 3 млн МЕ х 3 раза в неделю, после чего количество тромбоцитов снизилось до нормальных значений. С 2006 г. уровень транскрипта BCR-ABL1 не детектируется, а аллельная нагрузка мутации CALR c.1154_1155insTTGTC (p.K385fs*47) с момента обнаружения находится в диапазоне 41—48 % (рис. 7).
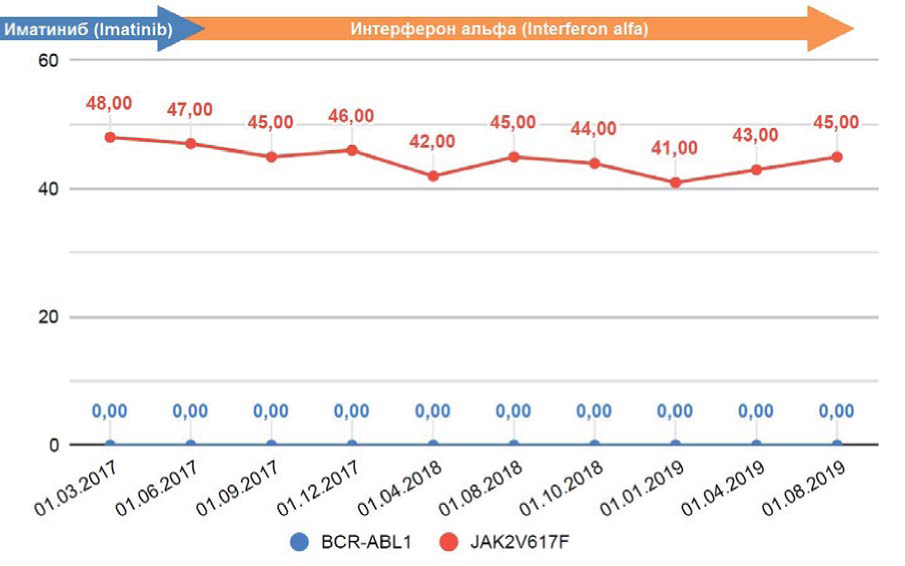
Рисунок 7. Кинетика уровней транскрипта BCR-ABLl и аллельной нагрузки CALR c.1154_1155insTTGTC p.K385fs*47 у больного В. В.С
Figure 7. Kinetics of the BCR-ABLl transcript level and allele burden of CALR c.1154_1155insTTGTC p.K385fs*47
По данным литературы, частота сочетания химерного гена BCR-ABL1 и мутации JAK2V617F у больных ХМЛ в различных популяциях широко варьирует: в США — 0,4 % [17][18], Германии — 0,2 % [19], Польше — 0,7 % [20], Мексике — 12,7 % [21], Пакистане — 26,7 % [22]. Сочетания химерного гена BCR-ABL1 и мутаций 9-го экзона CALR — относительно редкие явления, частота этих событий описана только для польской популяции больных ХМЛ и составляет 0,17 % [17]. В обследованной нами когорте больных ХМЛ частота сочетания химерного гена BCR-ABL1 с JAK2V617F составила 0,88 % (5/567), а с мутациями 9-го экзона гена CALR — 0,32 % (2/567). Медиана возраста на момент обнаружения сочетаний химерного гена BCR-ABL1 и мутации JAK2V617F в США составила 66 лет (диапазон 48—81 год) [18], в Германии — 72 года (диапазон 46—80 лет) [19]. Медиана возраста обследованных нами больных на момент обнаружения двух мутаций составила 60 лет (диапазон 51—66 лет). Анализ гендерных различий в случаях сочетания транскрипта BCR- ABL1 и JAK2V611F показал незначительное преобладание больных женского пола: от 52 % [19] до 64 % [18], тогда как в нашем исследовании большинство (4/7) составляли мужчины. При анализе литературных данных не удалось найти сведений о биологических закономерностях и хронологической последовательности приобретения химерного гена BCR-ABL1 и мутаций генов JAK2 и CALR. В представленном исследовании химерный ген BCR-ABL1 был выявлен ранее других мутаций в случаях 3, 6, 7; являлся вторым генетическим событием в случаях 2, 4, 5 и одновременно — в случае 1. Частота одномоментного выявления двух генетических аномалий среди больных в США составила 45 % (5/11), а в 55 % (6/11) случаев временной интервал был 87 месяцев (45—129 месяцев) [18]. В каждом случае ПМФ и ЭТ с мутацией JAK2V611F, а также в 3 случаях ИП с мутацией JAK2V611F в дальнейшем были выявлены химерный транскрипт BCR- ABL1 и развитие ХМЛ [18]. Подобный клинический феномен трансформации JAK2V611F+ ИП в BCR-ABL1+ ХМЛ через 12—18 лет наблюдения описан и другими исследователями [6][38].
Имеются данные о развитии JAK2V617F+ и CALR+ МПН у больных ХМЛ с цитогенетической и глубокой молекулярной ремиссией [3][16]. В описанном нами наблюдении № 7 CALR+ ЭТ был выявлен через 17 лет после установления клинического диагноза ХМЛ и при глубоком МО. Нерешенным остается вопрос, является ли химерный ген BCR-ABL1 и мутации генов JAK2V617F и CALR молекулярными событиями одной патогенетической цепи или нет. Результаты некоторых исследований показывают сложность молекулярного патогенеза МПН, зачастую не ограничивающегося тремя общепризнанными «драйверами». Известно, что в 47 % случаев «драйверным» мутациям предшествуют мутации генов DNMT5A, TET2, ASXL1, IDH2, SRSF2 и EZH2, вероятно, являющиеся предрасполагающими факторами геномной нестабильности гемопоэтической стволовой клетки (ГСК) [34][39][40]. Методом секвенирования следующего поколения (NGS — next generation sequencing) в образцах 23 больных с сочетанными мутациями BCR-ABL1 и JAK2V617F были обнаружены мутации в генах TET2, ASXL1, RUNX1, CBL, DNMT5A, PHF6, SF5B1 и TP55 с высокой частотой, что также может указывать на их роль в качестве предрасполагающих факторов в молекулярном патогенезе МПН [19].
Анализ литературы показал, что кинетика отдельных клонов, определяемая по изменению уровня транскрипта BCR-ABL1 и аллельной нагрузки JAK2V617F и CALRmun 1,2, имеет свои особенности. В немецком исследовании у 88 % (16/18) больных ХМЛ определялась разновекторная кинетика транскрипта BCR-ABL1 и аллельной нагрузки JAK2V617F, что, по мнению авторов, является результатом сосуществования двух разных клонов [19]. Синхронная кинетика уровня транскрипта BCR-ABL1 и аллельной нагрузки JAK2V617F под действием таргетной терапии препаратами ИТК наблюдается редко. Только у 18 % (2/11) больных ХМЛ в США отмечалось синхронное снижение уровня транскрипта BCR-ABL1 и аллельной нагрузки JAK2V617F до недетектируемого уровня [18].
В клиническом наблюдении 5 терапия иматинибом в дозе 400 мг/сут в течение 24 месяцев привела к синхронному снижению уровня транскрипта BCR-ABL1 с 1,77 до 0 %, а аллельной нагрузки мутации JAK2V617F — с 76 до 1 %. Учитывая, что препараты ИТК являются селективными ингибиторами только рецепторных тирозинкиназ ABL1, KIT и PDGF, но не активны в отношении JAK2, мы предполагаем, что в этом случае обе мутации возникли в одной ГСК, давшей начало всему патогенному клону МПН.
Какова прогностическая важность обнаружения сочетаний транслокации BCR-ABL1 с другими драйверными мутациями при МПН? Существует мнение, что мутация JAK2V617F может быть плохим прогностическим маркером ранней прогрессии ХМЛ [22]. Анализ собственных результатов показал, что лишь в 28 % (2/7) случаев ХМЛ с мутацией JAK2V617F не был достигнут БМО при терапии препаратами ИТК. При этом в 72 % (5/7) случаев больные достигли глубокого МО, а у 4 больных с глубоким МО терапия ИТК была прекращена, и больные сохраняют молекулярную ремиссию без таргетного лечения.
По каким признакам можно подозревать наличие второго МПН? Наши данные подтверждают описанные в литературе наблюдения, свидетельствующие о несоответствии выявляемой гепатомегалии и спленомегалии, нарастающих тромбоцитоза, лейкоцитоза или эритроцитоза с уровнем цитогенетического ответа или МО у больных ХМЛ, что является показанием к поиску других драйверных мутаций МПН [16]. В обследованной популяции больных ХМЛ наиболее частым признаком сопутствующего МПН были нарастающие лейкоцитоз (в случаях 2, 4, 5) и тромбоцитоз (в случаях 6 и 7), часто не соответствующие уровню транскрипта BCR-ABL1.
Не существует единой позиции и в вопросах формирования клинического диагноза и выбора терапии. Например, по мнению американских ученых, совместное появление транскрипта BCR-ABL1 и JAK2V617F чаще всего отражает два различных миелопролиферативных новообразования [18]. Клинико-фенотипическое доминировании одной из нозологий объясняется феноменом «пролиферативной конкуренции», согласно которой BCR-ABL1 является источником более сильной сигнализации, ведущей к более высокой пролиферации, чем мутация JAK2V611F или CALRmun 1,2 [14][22].
Терапевтический эффект ингибиторов BCR-ABL1- тирозинкиназ проявляется изолированным снижением уровня транскрипта BCR-ABL1, но не аллельной нагрузки мутаций генов JAK2 и CALR. В исследованной группе больных мы выделили три типа кинетики изменения уровни транскрипта BCR-ABL1 и аллельной нагрузки мутации JAK2V611F (рис. 8).

Рисунок 8. Клональная архитектура МПН и кинетика уровней транскрипта BCR- ABLl и аллельной нагрузки мутации JAK2V617F под воздействием терапии препаратами ИТК
Figure 8. Clonal architecture of MPN and kinetics of the BCR-ABLl transcript level and allele burden ofJAK2V6l7F under TKI therapy
Первый тип кинетики (параллельное снижению уровней транскрипта BCR-ABL1 и аллельной нагрузки мутации JAK2V611F), наблюдаемый при таргетной терапии препаратами ИТК, характерен когда один клон несет две мутации (патогенез МПН является бимутационно-моноклональной) (рис. 8 А). Подобное наблюдение также описано GR. Soderquist и соавт. [18]. Второй тип кинетики (вектор кинетики уровня транскрипта BCR- ABL1 и мутации JAK2V611F имеет разнонаправленный характер — снижение уровня BCR-ABL1 сопровождается ростом аллельной нагрузки JAK2V617F) характерен для МПН с двумя клонами, несущими в себе по одной мутации, как в описанном клиническом наблюдении 2 (рис. 8 В). В данном случае элиминация BCR-ABL1 клона под воздействием ИТК приводит к расширению JAK2V611F+ клона и фенотипической манифестации сопутствующего МПН [16]. Третий тип кинетики (аллельная нагрузка JAK2V611F сначала синхронно снижается с уровнем BCR-ABL1, а затем их кинетика становится разнонаправленной) характерен для патогенеза МПН со сложной клональной архитектурой. Вероятно, снижение аллельной нагрузки JAK2V611F при терапии препаратом ИТК происходит за счет элиминации JAK2V611F+/ BCR-ABL1+ клона, как в случаях 1 и 5 (рис. 8 С). К аналогичному выводу приходят также J. Grisouard и соавт. [12], описывающие случай, когда из трех (BCR-ABL1+, JAK2V611F+ и BCR-ABL1+/JAK2V6nFF) клонов наиболее чувствительным к терапии препаратами ИТК оказался BCR-ABL1+/JAK2V611F+ субклон. Вопрос о необходимости описания и классификации миелопролиферативных новообразований с более чем одной генетической аномалией уже поднимался в литературе [16]. Мы уверены, что дальнейшее накопление сведений о патогенетических механизмах сочетаний разных нозологий МПН необходимо для выработки адекватных клинических диагнозов и разработки эффективных методов комбинированной таргетной терапии с включением препаратов, направленных на сопутствующие драйверные мутации.
1. Swerdlow S.H., Campo E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H. et al. (Eds): WHO Classifi cation of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th edition). IARC: Lyon, 2017.
2. Mirza I., Frantz C., Clarke G. et al. Transformation of polycythemia vera to chronic myelogenous leukemia. Arch Pathol Lab Med. 2007; 131(11): 1719–24. DOI: 10.1043/1543-2165(2007)131[1719: TOPVTC]2.0.CO;2.
3. Inami M., Inokuchi K., Okabe M. et al. Polycythemia associated with the JAK2V617F mutation emerged during treatment of chronic myelogenous leukemia. Leukemia. 2007; 21(5): 1103–1104. DOI: 10.1038/sj.leu.2404591.
4. Bee P.C., Gan G.G., Nadarajan V.S. et al. A man with concomitant polycythaemia vera and chronic myeloid leukemia: the dynamics of the two disorders. Int J Hematol. 2010; 91: 136–139. DOI: 10.1007/s12185-009-0471-6.
5. Cambier N., Renneville A., Cazaentre T. et al. JAK2V617F-positive polycythemia vera and Philadelphia chromosome-positive chronic myeloid leukemia: one patient with two distinct myeloproliferative disorders. Leukemia. 2008; 22(7): 1454–5. DOI: 10.1038/sj.leu.2405088.
6. Wang X., Tripodi J., Kremyanskaya M. et al. BCR-ABL1 is a secondary event after JAK2V617F in patients with polycythemia vera who develop chronic myeloid leukemia. Blood. 2013; 121(7): 1238–9. DOI: 10.1182/blood-2012-11-467787.
7. Siricilla M., Nader K., Ferber A. A case report of chronic myelogenous leukemia with JAK2- and BCR-ABL-positive mutation. Am J Hematol Oncol. 2017; 13(2).
8. Qin Y.W., Yang Y.N., Li S. et al. Coexistence of JAK2V617F mutation and BCRABL translocation in a pregnant woman with essential thrombocythemia. Indian J Hematol Blood Transfus. 2014; 30(suppl 1): 331–4. DOI: 10.1007/s12288-014- 0385-1.
9. Wahlin A., Golovleva I. Emergence of Philadelphia positive chronic myeloid leukemia during treatment with hydroxyurea for Philadelphia negative essential thrombocytosis. Eur J Hematol. 2003; 70(4): 240–1. DOI: 10.1034/j.1600- 0609.2003.00043.x.
10. Lippert E., Boissinot M., Kralovics R. et al. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood. 2006; 108(6): 1865–7. DOI: 10.1182/blood-2006-01-013540.
11. Tabassum N., Saboor M., Ghani R. et al. Frequency of JAK2 V617F mutation in patients with Philadelphia positive chronic myeloid leukemia in Pakistan. Pak J Med Sci. 2014; 30(1): 185–8. DOI: 10.12669/pjms.301.3906.
12. Grisouard J., Ojeda-Uribe M., Looser M. et al. Complex subclone structure that responds differentially to therapy in a patient with essential thrombocythemia and chronic myeloid leukemia. Blood. 2013; 122(22): 3694–6. DOI: 10.1182/ blood-2013-07-516385.
13. Hassankrishnamurthy S., Mody M.D., Kota V.K. A Case of Chronic Myelogenous Leukemia Occurring in a Patient Treated for Essential Thrombocythemia. Am J Case Rep. 2019; 20: 10–4. DOI: 10.12659/AJCR.911854.
14. Hussein K., Bock O., Seegers A. et al. Myelofi brosis evolving during imatinib treatment of a chronic myeloproliferative disease with coexisting BCR-ABL translocation and JAK2V617F mutation. Blood. 2007; 109(9): 4106–7. DOI: 10.1182/ blood-2006-12-061135.
15. Bader G., Dreiling B. Concurrent JAK2-Positive Myeloproliferative Disorder and Chronic Myelogenous Leukemia: A Novel Entity? A Case Report with Review of the Literature. J Investig Med High Impact Case Rep. 2019; 7: 1–5. DOI: 10.1177/2324709619832322.
16. Lee Y.J., Moon J.H., Shin H.C. et al. Two CML patients who subsequently developed features of essential thrombocythemia with JAK2-V617F mutation while in complete cytogenetic remission after treatment with imatinib mesylate. Int J Hematol. 2013; 97(6): 804–7. DOI:10.1007/s12185-013-1326-8.
17. Jelinek J., Oki Y., Gharibyan V. et al. JAK2 Mutation 1849G>T is Rare in Acute Leukemias but Can be Found in CMML, Philadelphia Chromosome negative CML, and Megakaryocytic Leukemia. Blood. 2005; 106(10): 3370–3. DOI: 10.1182/blood-2005-05-1800.
18. Soderquist C.R., Ewalt M.D., Czuchlewski D.R. et al. Myeloproliferative neoplasms with concurrent BCR-ABL1 translocation and JAK2 V617F mutation: a multiinstitutional study from the bone marrow pathology group. Mod Pathol. 2018; 31(5): 690–704. DOI: 10.1038/modpathol.2017.182
19. Martin-Cabrera P., Haferlach C., Kern W. et al. BCR-ABL1-positive and JAK2 V617F-positive clones in 23 patients with both aberrations reveal biologic and clinical importance. Br J Hematol. 2017; 176(1): 135–9. DOI: 10.1111/bjh.13932.
20. Lewandowski K., Wojtaszewska M., Kanduіa Z. et al. Coexistence of JAK2 or CALR mutation is a rare but clinically important event in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Int J Lab Hematol. 2018; 40(3): 366–71. DOI: 10.1111/ijlh.12798.
21. Trejo R.M.A., Gonzalez V.A., Saldivar I. et al. High Frequency of Concurrent JAK2 V617F Mutation and BCR/ABL Fussion Gene in a Cohort (18/142) of Mexican Patients with MPD. Blood. 2012; 120: 1766.
22. Pahore Z.A., Shamsi T.S., Taj M. et al. JAK2V617F mutation in chronic myeloid leukemia predicts early disease progression. J Coll Physicians Surg Pak. 2011; 21(8): 472–5. DOI: 08.2011/JCPSP.472475.
23. Nangalia J., Massie C.E., Baxter E.J. et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013; 369: 2391–405. DOI: 10.1056/NEJMoa1312542.
24. Klamfl T., Gisslinger H., Harutyunyan A.S. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013; 369: 2379–90. DOI: 10.1056/NEJMoa1311347.
25. Qiao C., Sun C., Ouyang Y. et al. Clinical importance of different calreticulin gene mutation types in wild-type JAK2 essential thrombocythemia and myelofi brosis patients. Haematologica. 2014; 99: e183. DOI: 10.3324/haematol.2014.109199.
26. Pagoni M., Garofalaki M., Tziotziou I. et al. Concurrent or Sequential BCRABL1 translocation and Calr gene or JAK2V617F mutation. 58th ASH Annual meeting. San Francisco. 6–9.12.2014. https://ash.confex.com/ash/2014/webprogram/Paper68049.html.
27. Cabagnols X., Cayuela J.M., Vainchenker W. A CALR Mutation Preceding BCR-ABL1 in an Atypical Myeloproliferative Neoplasm. N Engl J Med. 2015; 372(7): 688–90. DOI: 10.1056/NEJMc1413718.
28. Bonzheim I., Mankel B., Klapthor P. et al. CALR-mutated essential thrombocythemia evolving to chronic myeloid leukemia with coexistent CALR mutation and BCR-ABL translocation. Blood. 2015; 125(14): 2309–11. DOI: 10.1182/ blood-2014-12-616847.
29. Loghavi S., Pemmaraju N., Kanagal-Shamanna R. et al. Insights from response to tyrosine kinase inhibitor therapy in a rare myeloproliferative neoplasm with CALR mutation and BCR-ABL1. Blood. 2015; 125(21): 3360–3. DOI: 10.1182/ blood-2015-03-632893.
30. Seghatoleslami M., Ketabchi N., Ordo A. et al. Coexistence of p190 BCR/ ABL Transcript and CALR 52-bp Deletion in Chronic Myeloid Leukemia Blast Crisis: A Case Report. Mediterr J Hematol Infect Dis. 2016; 8(1): e2016002. DOI: 10.4084/MJHID.2016.002.
31. Diamond J.M., de Almeida A.M., Belo H.J. et al. CALR-mutated primary myelofi brosis evolving to chronic myeloid leukemia with both CALR mutation and BCR-ABL1 fusion gene. Ann Hematol. 2016; 95(12): 2101–4. DOI: 10.1007/ s00277-016-2827-3.
32. Patel S., Kim S.H., Shammo J.M. et al. Patient with myeloproliferative neoplasms (MPN) who later develop ph+ chronic myelogenous leukemia (CML): a case series. J Clin Oncol. 2017; 35(suppl): e18563. DOI: 10.1200/JCO.2017.35.15_ suppl.e18563.
33. Dogliotti I., Carmen F., Serra A. et al. CALR-positive myeloproliferative disorder in a patient with Ph-positive chronic myeloid leukemia in durable treatmentfree remission: a case report. Stem Cell Investig. 2017; 4: 57. DOI: 10.21037/ sci.2017.06.02.
34. Kandarpa M., Wu Y.M., Robinson D. et al. Clinical characteristics and whole exome/transcriptome sequencing of coexisting chronic myeloid leukemia and myelofi brosis. Am J Hematol. 2017; 92(6): 555–61. DOI: 10.1002/ajh.24728.
35. De Roeck L., Michaux L., Debackere K. et al. Coexisting driver mutations in MPN: clinical and molecular characteristics of a series of 11 patients. Hematology. 2018; 11: 1–8. DOI: 10.1080/10245332.2018.1498182.
36. Boddu P., Chihara D., Masarova L. et al. The co-occurrence of driver mutations in chronic myeloproliferative neoplasms. Ann Hematol. 2018; 97(11): 2071–80. DOI: 10.1007/s00277-018-3402-x.
37. Mughal T.I., Gotlib J., Mesa R. et al. Recent advances in the genomics and therapy of BCR/ABL1-positive and -negative chronic myeloproliferative neoplasms. Leuk Res. 2018; 67: 67–74. DOI: 10.1016/j.leukres.2018.02.008.
38. Pingali S.R., Mathiason M.A., Lovrich S.D., Go R.S. Emergence of chronic myelogenous leukemia from a background of myeloproliferative disorder: JAK2V617F as a potential risk factor for BCR-ABL translocation. Clin Lymphoma, Myeloma. 2009; 9(5): E25–9. DOI: 10.3816/CLM.2009.n.080.
39. Magor G.W., Tallack M.R., Klose N.M. et al. Rapid Molecular Profi ling of Myeloproliferative Neoplasms Using Targeted Exon Resequencing of 86 Genes Involved in JAK-STAT Signaling and Epigenetic Regulation. J Mol Diagn. 2016; 18: 707–18. DOI: 10.1016/j.jmoldx.2016.05.006.
40. Stein B.L., Williams D.M., O’Keefe C. et al. Disruption of the ASXL1 gene is frequent in primary, post-essential thrombocytosis and post-polycythemia vera myelofi brosis, but not essential thrombocytosis or polycythemia vera: analysis of molecular genetics and clinical phenotypes. Haematologica. 2011; 96: 1462–9. DOI: 10.3324/haematol.2011.045591.
Треглазова Светлана Анатольевна, научный сотрудник лаборатории молекулярной гематологии
Горячева Светлана Рудольфовна, кандидат медицинских наук, врач-гематолог консультативного гематологического отделения с дневным стационаром по проведению интенсивной высокодозной химиотерапии
Абдуллаев А.О., Степанова Е.А., Макарик Т.В., Никулина Е.Е., Треглазова С.А., Горячева С.Р., Шухов О.А., Быкова А.В., Трацевская Ж.В., Меликян А.Л., Ковригина А.М., Туркина А.Г., Судариков А.Б. Частота сочетания и кинетика уровня транскрипта BCR-ABL1 и аллельной нагрузки мутаций JAK2V617F+ и CALR тип-1, -2 у больных хроническим миелолейкозом. Гематология и трансфузиология. 2020;65(3):253-280. https://doi.org/10.35754/0234-5730-2020-65-3-263-280
Abdullaev A.O., Stepanova E.A., Makarik T.V., Nikulina E.Y., Treglazova S.A., Goryacheva S.R., Shukhov O.A., Bykova A.V., Tratsevskaya Z.V., Melikyan A.L., Kovrigina A.M., Turkina A.G., Sudarikov A.B. Frequency of coexistence and kinetics of the BCR-ABL1 transcript level and allele burden of JAK2V617F and CALR Type 1, 2 gene mutations in patients with chronic myeloid leukemia. Russian journal of hematology and transfusiology. 2020;65(3):253-280. (In Russ.) https://doi.org/10.35754/0234-5730-2020-65-3-263-280
![]()
125167, Москва, Новый Зыковский проезд, 4
ФГБУ «НМИЦ гематологии» Минздрава России
тел.: 8-926-816-3887
e-mail: o.levchenko@htjournal.ru






































