


Содержание
Перейти к:
https://doi.org/10.35754/0234-5730-2021-66-3-322-345
Перейти к:
Введение. Минорные антигены гистосовместимости (МАГ) — полиморфные пептиды, презентируемые в молекулахHLA, являются продуктами генов, содержащих несинонимичные однонуклеотидные полиморфизмы. При трансплантации аллогенных гемопоэтических стволовых клеток (алло-ТГСК) иммунный ответ, направленный на МАГ, можетприводить как к реакции «трансплантат против хозяина», так и к реакции «трансплантат против опухоли». Некоторые МАГ представляют собой перспективные и безопасные мишени для Т-клеточной иммунотерапии рецидивов лейкозов после алло-ТГСК.
Цель — анализ литературы, описывающей иммунный ответ на различные МАГ, а также клинические исследования, использующие МАГ как мишень иммунотерапии.
Основные сведения. МАГ являются перспективными мишенями для профилактики или терапии рецидивов лейкозов после алло-ТГСК за счет того, что они обладают преимуществами по сравнению с другими классами мишеней:опухоль-ассоциированными антигенами и неоантигенами. Для того, чтобы быть пригодными для иммунотерапии, МАГ должен удовлетворять ряду параметров: 1) презентироваться распространенным аллелем HLA, 2) иметь оптимальное соотношение частот аллельных вариантов кодирующего полиморфизма, 3) кодироваться геном, преимущественно экспрессирующимся в кроветворной ткани. Это резко ограничивает число применимых мишеней и делает актуальным поиск новых МАГ.
Пилунов А.М., Романюк Д.С., Ефимов Г.А., Савченко В.Г. Минорные антигены гистосовместимости как мишени Т-клеточной иммунотерапии. Гематология и трансфузиология. 2021;66(3):322-345. https://doi.org/10.35754/0234-5730-2021-66-3-322-345
Pilunov A.M., Romaniuk D.S., Efimov G.A., Savchenko V.G. Minor histocompatibility antigens as targets for T-cell immunotherapy. Russian journal of hematology and transfusiology. 2021;66(3):322-345. (In Russ.) https://doi.org/10.35754/0234-5730-2021-66-3-322-345
Трансплантация аллогенных гемопоэтических стволовых клеток (алло-ТГСК) является «золотым стандартом» терапии острых миелоидных (ОМЛ) и лимфобластных (ОЛЛ) лейкозов [1]. Однако приблизительно у половины больных после алло-ТГСК развивается рецидив заболевания [2–6], что делает актуальной разработку методов терапии рецидивов [7–9]. Терапевтический эффект алло-ТГСК в значительной степени обусловлен реакцией «трансплантат против лейкоза» (РТПЛ), при которой лимфоциты донора уничтожают злокачественные клетки реципиента, пережившие кондиционирование [10–12]. В то же время аллореактивные лимфоциты донора могут атаковать здоровые ткани реципиента, приводя к реакции «трансплантат против хозяина» (РТПХ) [13–15]. При алло-ТГСК, при которой донор и реципиент не полностью совместимы по лейкоцитарным антигенам человека (Human Leukocyte Antigen, HLA), иммунный ответ развивается за счет донорских Т-лимфоцитов, которые распознают клетки реципиента, несущие аллогенные аллели HLA [16]. Такое распознавание обусловлено кросс-реактивностью Т-лимфоцитов, содержащихся в популяции клеток памяти [17]. Однако иммунный ответ может возникать и при полностью HLA-совместимой алло-ТГСК, при которой мишенью иммунного ответа являются минорные антигены гистосовместимости (МАГ) [18–20] — полиморфные пептиды, презентируемые в молекулах HLA, которые являются продуктами генов, содержащих несинонимичные однонуклеотидные полиморфизмы (нсОНП). При отсутствии у донора одной из аллельных форм пептида к ней не развивается иммунологическая толерантность, соответственно, среди всего разнообразия донорских лимфоцитов могут встречаться те, которые имеют Т-клеточные рецепторы (ТКР), специфичные к данному пептиду. При попадании таких лимфоцитов в организм реципиента, имеющего иммунологически чужеродную аллельную форму пептида, может возникать аллогенный иммунный ответ. Т-лимфоциты, специфичные к МАГ, преимущественно встречаются среди популяции наивных клеток [21].
Аллогенный иммунный ответ имеет двойственный эффект: с одной стороны, развитие РТПЛ способствует благоприятному исходу алло-ТГСК, с другой, — РТПХ поражает здоровые ткани организма и является потенциально жизнеугрожающим осложнением алло-ТГСК. Долгое время оставался открытым вопрос, насколько взаимосвязаны эти явления. По данным исследований [22–24], у больных с острой или хронической РТПХ рецидивы ОМЛ и ОЛЛ возникают значительно реже. При трансплантации аутологичных или сингенных стволовых клеток, при которой отсутствует риск развития РТПХ, вероятность рецидива заболевания существенно выше, чем при алло-ТГСК, что связано с отсутствием РТПЛ [23][25]. Более поздние исследования показали зависимость между возникновением РТПХ и степенью генетического различия пары «донор — реципиент». Ретроспективный анализ 32 828 больных, которым была выполнена алло-ТГСК, в котором напрямую учитывались различия по HLA и косвенно — по минорным антигенам, показал уменьшение летальности, не связанной с рецидивом заболевания, при уменьшении различий между донором и реципиентом [26]. Позитивный терапевтический эффект алло-ТГСК, выраженный в безрецидивной выживаемости, не зависел от степени различия пар «донор — реципиент» по HLA. Это свидетельствует, что РТПЛ может быть отделена от РТПХ, а МАГ можно использовать в качестве мишеней иммунного ответа для профилактики рецидивов [27].
Цель настоящего обзора — анализ литературы, описывающей иммунный ответ на различные МАГ, а также клинические испытания, использующие МАГ как мишень иммунотерапии.
Впервые МАГ описаны в 1975 г. [28]. Было установлено, что иммунизация мышей гемопоэтическими клетками, полученными от другой линии мышей, идентичной по локусу главного комплекса гистосовместимости (ГКГС), но при этом отличающейся по другим генам, приводит к иммунному ответу. Цитотоксичность полученных линий Т-лимфоцитов подтверждена in vitro. Был сделан вывод, что существуют некие антигены гистосовместимости, не закодированные в локусе ГКГС. Распознавание этих антигенов обеспечивается Т-лимфоцитами, деплеция Т-лимфоцитов перед трансплантацией позволяет избежать летальности у мышей-реципиентов [29][30]. Одной из важнейших вех стало открытие функции молекул ГКГС в презентации пептидных антигенов Т-лимфоцитам, как чужеродных, так и происходящих из собственных белков организма [31]. Было показано, что МАГ представляют собой презентируемые в контексте ГКГС I аутологичные пептиды, различающиеся по аминокислотной последовательности у донора и реципиента за счет несинонимичных полиморфизмов [32].
Возникновение РТПХ у больных после полностью HLA-совместимой алло-ТГСК подтвердило наличие антигенов гистосовместимости, отличных от HLA и у человека [33, 34]. За счет различия по МАГ вероятность возникновения РТПХ при трансплантации от неродственного HLA-совместимого донора выше, чем при трансплантации от HLA-идентичного сиблинга [35]. Аллореактивные Т-клетки донора способны распознавать различия в одну единственную аминокислоту в последовательности пептида и инициировать иммунный ответ [36]. Для иммунной системы донора отсутствующие в его организме аллели минорных антигенов являются чужеродными, специфичные к ним ТКР не подвергаются негативной селекции в тимусе и толеризации на периферии, в отличие от Т-клеток, специфичных к аутологичным аллельным вариантам, что было показано в экспериментальной модели на мышах [37]. Таким образом, у доноров, не имеющих конкретного аллеля минорного антигена, могут встречаться Т-лимфоциты, специфически распознающие кодируемый им пептид [38].
Был описан [39] первый аутосомный минорный антиген человека, который был назван НА. Несоответствие по этому антигену привело к развитию РТПХ, возникшей после трансплантации от HLA-совместимой сестры к брату. HA антиген не был связан с Y-хромосомой, группой крови, резус-фактором и белками комплемента. Установлено, что этот антиген презентируется в наиболее распространенном у европейцев аллеле HLA — HLA-A2*02. Полученные клоны цитотоксических Т-лимфоцитов распознавали гемопоэтические клетки большинства доноров, несущих этот аллель HLA, что свидетельствовало о высокой частоте встречаемости антигена HA в популяции. В дальнейшем были обнаружены другие минорные антигены человека, как связанные с Y-хромосомой, так и аутосомные [40]. Анализ иммунного ответа in vitro цитотоксических клонов, специфичных к 5 МАГ (НА-1–НА-5), на клетки здоровых доноров показал, что МАГ встречаются с различной частотой: например, антигены HA-2 и НА-3 встречались повсеместно в исследованной популяции, в то время как НА-4 и НА-5 были очень редки. Частота встречаемости антигена НА-1 была около 70 %. Частота обнаружения цитотоксических Т-лимфоцитов, специфичных к HA-1, после несовместимой по этому антигену алло-ТГСК была существенно больше, чем для других антигенов. Авторы пришли к выводу, что существует феномен иммунодоминантности МАГ, а НА-1 является наиболее иммуногенным из изученных антигенов, за счет чего при несовпадении между донором и реципиентом по НА-1 иммунный ответ направлен преимущественно на этот антиген [41].
Изучение специфичности цитотоксических клонов, полученных от больных с РТПХ, подтвердило феномен иммунодоминантности: при HLA-несовместимой трансплантации цитотоксические клоны обладали специфичностью к несовместимой молекуле HLA, при полностью HLA-совместимой трансплантации, несовместимой по НА-1, все выделенные аллореактивные клоны были специфичны к НА-1, а при совместимости по HLA и НА-1 иммунный ответ развивался на антигены Y-хромосомы [42]. Таким образом, иммунный ответ на аллоантигены иерархичен: подавляющее большинство образующихся аллореактивных клонов цитотоксических Т-лимфоцитов специфично к наиболее иммунодоминантному антигену, на субдоминантные антигены в таком случае иммунный ответ практически не развивается. Иерархичность иммунного ответа на МАГ подтверждена у мышей [43]. Установлено, что Т-клеточные клоны, специфичные к иммунодоминантным МАГ, способны уничтожать клетки лейкоза у мышей, в то время как клоны, специфичные к субдоминантным МАГ, не способны оказать противоопухолевого действия [44]. Число иммунодоминантных антигенов невелико: примерно 0,5 % от пептидов, теоретически способных связаться с HLA (Kd < 500 nM), обладают достаточной аффинностью для иммунодоминантности (Kd < 50 nM), однако из-за неэффективного процессинга антигена, а также отсутствия в организме ТКР к некоторым пептидным антигенам, количество иммунодоминантных пептидов оценивается еще ниже, примерно в 0,2 % от потенциально презентируемых пептидов [45].
Один из механизмов иммунодоминантности — конкуренция пептидов за связывание с HLA. Данный феномен наблюдается, только если доминантный и субдоминантный антиген экспрессируется в одном типе клеток [46] и презентируются одним и тем же аллельным вариантом HLA. Иммунодоминантным в таком случае является антиген, который имеет наибольшую аффинность связывания с HLA [47, 48]. Другой возможный механизм возникновения иммунодоминантности МАГ — сильно различающаяся частота встречаемости специфичных ТКР из-за различной вероятности их образования в процессе соматической рекомбинации. Это было показано на примере мышиного минорного антигена B6dom1, который был более иммунодоминантным, чем Н3 и Н13, хотя аффинность связывания с ГКГС этих антигенов приблизительно одинакова [49]. Была проведена оценка разнообразия бета-цепей ТКР, специфичных к этим МАГ, среди наивных Т-клеток мышей, не имевших аллеля МАГ. Выяснилось, что разнообразие B6dom1-специфичных ТКР бета-цепей значительно выше.
Иммунодоминантность МАГ наблюдается и в клинической практике. В исследовании [50] были проанализированы 327 HLA-совместимых ТГСК. Пары «донор — реципиент» были генотипированы по 17 МАГ, презентирующимся в 6 разных аллельных вариантах HLA. Исследование подтвердило, что несовпадение по аутосомным МАГ ассоциировано с более низкой частотой рецидива как после трансплантации от родственного, так и неродственного донора. Установлена корреляция между несовпадением по Y-хромосомным МАГ и возникновением РТПХ, при этом подобная корреляция для аутосомных МАГ не установлена. О роли МАГ-специфичных лимфоцитов в развитии РТПЛ свидетельствует тот факт, что у 10–33 % реципиентов с помощью окраски HLA-тетрамерами в крови были обнаружены донорские МАГ-специфичные клетки, при этом их наличие было ассоциировано с лучшей безрецидивной выживаемостью. Однако иммуногенность МАГ сильно различалась. CD8+ Т-клетки, специфичные к НA1, HA2, PANE1, LRH1, ACC1, и к Y-хромосомным МАГ HY.A2 и HY.B7, были обнаружены у 25–60 % реципиентов с несовместимостью по МАГ. HA8, SP110, и ZAPHIR вызывали иммунный ответ у 10–20 % реципиентов. В этом исследовании T-клеточный ответ на МАГ ADIR, HwA11, ECGF, HEATR и HY.B8 не был обнаружен, несмотря на генетические различия. Таким образом, не все существующие МАГ одинаково способны вызывать иммунный ответ, и нет однозначных методов предсказания их иммуногенности; детекция иммунного ответа in vivo — единственный надежный критерий [51].
Существует несколько механизмов возникновения МАГ. Первые описанные МАГ — это производные генов, закодированных на Y-хромосоме: в женском организме эти гены отсутствуют, и при алло-ТГСК от донора-женщины реципиенту-мужчине возможно их распознавание и развитие РТПХ [52]. Однако большая часть описанных МАГ кодируется полиморфизмами в аутосомных генах (рис. 1). Трансляция белков сопровождается образованием определенной доли дефектных рибосомальных продуктов [53] по нескольким механизмам: трансляция пре-сплайсированной мРНК [54], трансляция мРНК, подвергающейся деградации при регуляторной РНК-интерференции [55] или трансляции некодирующих участков генома [56]. Дефектные рибосомальные продукты, а также неправильно свернутые белки и белки с истекшим жизненным сроком убиквитинилируются [57], а затем расщепляются протеасомой на пептиды длиной 4–20 аминокислот [58][59]. Полученные пептиды транспортируются белком-транспортером, связанным с процессингом антигена (Transporter associated with antigen processing, TAP) в эндоплазматический ретикулум [60], где они могут связываться с одним из аллельных вариантов моолекулы HLA, а затем презентироваться на поверхности клетки [61]. Если презентируемый пептид содержит полиморфную аминокислоту, образуется потенциальный МАГ. При этом возможны два различных варианта:
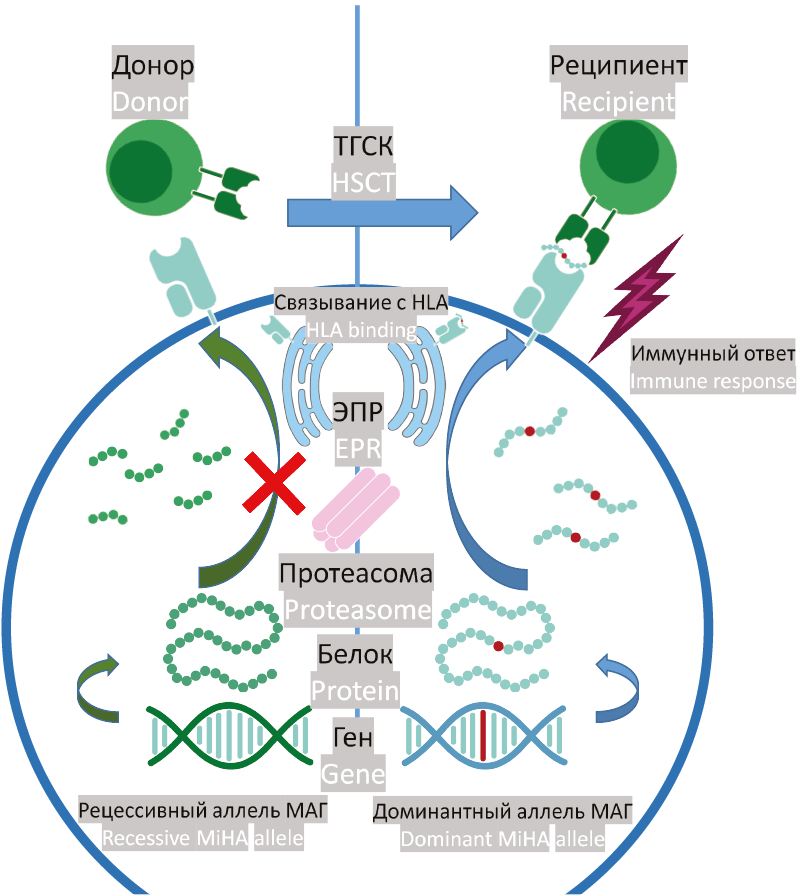
Рисунок 1. Образование МАГ по доминантно-рецессивному механизму. Белок, содержащий полиморфизм (отмечен красным), процессируется протеасомой на пептиды, которые транспортируются в эндоплазматический ретикулум, где они связываются с HLA, а затем представляются на поверхности клетки. Нарушения на какой-либо из этих стадий приводят к тому, что пептид из рецессивного аллеля МАГ отсутствует на поверхности клетки. Лимфоциты донора, не имеющего МАГ, способны распознать клетки реципиента, имеющего МАГ
Figure 1. Dominant-recessive mechanism of MiHA generation. The protein containing the polymorphism (marked in red) is processed by the proteasome into peptides, which are transported to the endoplasmic reticulum where they bind to HLA molecules and then are exposed on the cell surface. Lymphocytes from a MiHA-negative donor are able to recognize MiHA-presenting recipient cells
Подавляющее большинство минорных антигенов доминантно-рецессивные, доля кодоминантных МАГ оценивается в 7 % [62]. Аминокислотная замена может повлиять на любой из этапов вывода пептида на поверхность клетки. Альтернативный аллельный вариант минорного антигена НА-3 расщепляется протеасомой на более мелкие фрагменты [63], альтернативный аллель НА-8 не взаимодействует с транспортером TAP [64], а альтернативный пептид НА-1 не презентируется за счет того, что он не обладает достаточной аффинностью связывания с HLA-A*02:01 [65].
По результатам анализа публично доступных геномных данных (проект «1000 геномов»), 90 % МАГ происходят из несинонимических однонуклеотидных полиморфизмов при сохранении функциональности обоих аллельных вариантов исходного белка [62]. Однако существуют и другие механизмы: минорный антиген может возникнуть, если один из аллелей гена содержит инсерцию/делецию (LRH-1) [66] или дополнительный стоп-кодон (PANE1) [67]; имеет нарушение в сплайсинге, приводящее к трансляции интрона (ZAPHIR) [68], делецию экзона (ACC-6) [69] и, наконец, в результате делеции всего гена (UGT2B17/A2) [70]. Детально генетические механизмы формирования МАГ описаны в обзоре [71]. В отдельных случаях иммуногенностью может обладать только пептид, имеющий определенную посттрансляционную модификацию, что было показано на примере цистеинилированного пептида белка SMCY из Y-хромосомы [72]. Показано [73], что 0,5 % всех нсОНП, различных между двумя сиблингами, приводили к тому, что вариантный пептид был представлен в иммунопептидоме, в то время как иммуногенностью обладали 0,22 %. При этом отсутствие иммунного ответа на один из аллельных вариантов МАГ не всегда объясняется тем, что пептид не презентируется в HLA. Исследовались [74] три МАГ: LB-CLYBL-1Y, LB-NISCH-1A и LB-SSR1–1S, считающихся доминантно-рецессивными. С помощью оценки связывания меченых пептидов с HLA на поверхности клеточной линии Т2 (неспособной презентировать эндогенные пептиды) показано, что для всех антигенов аффинность связывания с HLA обоих аллельных вариантов пептидов идентична. При этом клоны Т-лимфоцитов, специфичные к иммуногенному аллельному варианту, не распознавали альтернативный пептид. Таким образом, отсутствие иммуногенности в этом случае объясняется, наиболее вероятно, отсутствием у доноров Т-лимфоцитов, способных распознавать эти антигены, что может быть результатом негативной тимусной селекции.
Большинство описанных МАГ презентируется в HLA I класса, однако минорные антигены могут презентироваться и в HLA II класса за счет презентации аутологичных пептидов, захваченных извне. Описано [75] возникновение РТПЛ, мишенью которой были минорные антигены, презентируемые в HLA II класса. С одной стороны, HLA II класса экспрессируются преимущественно в гемопоэтических клетках, и иммунный ответ на презентируемые ими МАГ может вызывать селективную РТПЛ в отсутствие РТПХ. С другой стороны, экспрессия HLA II класса в опухолевых клетках часто подавляется как механизм ускользания от иммунного ответа [76, 77], за счет того, что потеря HLA-II, в отличие от потери HLA-I, не вызывает цитотоксического ответа со стороны NK-клеток [78]. В качестве мишеней для иммунотерапии представляют интерес скорее минорные антигены, презентируемые в HLA I класса.
Биоинформатический анализ геномов предсказывает тысячи полиморфизмов, способных кодировать минорные антигены, представляемые в наиболее частых аллелях HLA I класса [56, 62]. В настоящее время описано относительно небольшое их количество — в обзоре [71] упоминаются 48 минорных антигенов HLA I класса и 8 минорных антигенов HLA II класса. Исследование В-лимфобластоидных линий с секвенированным геномом из проекта «1000 геномов» увеличило количество известных МАГ I класса до 63 [79].
Поиск новых минорных антигенов методами «прямой иммунологии» — через выявление мишеней естественного возникающего иммунного ответа — затруднен за счет феномена иммунодоминантности [42]. При HLA-совместимой неродственной алло-ТГСК степень различия по МАГ в 2 раза больше, чем при HLA-совместимой трансплантации от сиблингов, однако она несет значительно меньший риск возникновения РТПХ, чем единственное несовпадение по одному из аллелей HLA-DP [80, 81]. Хотя количество несовпадений по несинонимичным полиморфизмам напрямую не коррелировало с тяжестью РТПХ, возникновение в 60 % случаев острой РТПХ и в 40 % случаев хронической РТПХ после трансплантации от родственного HLA-совместимого донора вызвано ответом на МАГ [80][81]. При трансплантации от полностью HLA-совместимого донора иммунный ответ ограничивается небольшим количеством наиболее иммунодоминантных МАГ, а различия по субдоминантным МАГ никак не проявляются.
Методы «обратной иммунологии» — с помощью проверки иммуногенности предсказанных МАГ — также оказываются не очень результативными. Полученные in vitro Т-клеточные клоны, распознающие экзогенные пептиды, предсказанных МАГ, не были способны распознать эндогенный пептид во всех случаях, кроме уже открытого МАГ НА-1 [82]. После открытия 50 терапевтически значимых МАГ для 35 % алло-ТГСК становится возможным использование МАГ-специфичной терапии [83].
Свидетельства роли МАГ в развитии РТПЛ подтолкнули к идее использования МАГ-специфичного иммунного ответа для разработки метода терапии лейкозов, не приводящего к развитию РТПХ [27][33]. Наиболее перспективными мишенями являются МАГ, происходящие из генов преимущественно или исключительно экспрессирующихся в гемопоэтической ткани, что позволит исключать вероятность развития РТПХ [84]. Показано [85], что МАГ-специфичные CD8+ Т-клоны, выделенные от больных с РТПЛ, но без РТПХ, обладают сниженной способностью распознавать негемопоэтические клетки реципиентов, по сравнению с лимфоцитами, полученными от больных с РТПХ. При этом количество тканеспецифичных МАГ крайне невелико: из 6000 проанализированных потенциальных МАГ лишь 39 специфично экспрессированы в гемопоэтической ткани [86].
МАГ обладают рядом преимуществ по сравнению с другими мишенями для терапии лейкозов [87]. МАГ представляют собой антигены, ранее не встречавшиеся с иммунной системой донора, и потому потенциально обладают большей иммуногенностью, чем опухоль-ассоциированные антигены — аберрантно экспрессированные в злокачественных клетках аутологичные белки. Таким образом, по иммуногенности МАГ соотносятся с опухолевыми неоантигенами — продуктами опухоль-специфичных мутаций в белок-кодирующих регионах, а также вирусными антигенами. По сравнению с неоантигенами, МАГ обладают тем преимуществом, что, во-первых, презентируются на всех опухолевых клетках, независимо от образования субклонов; во-вторых, не являются уникальными для конкретного больного. Следовательно, МАГ-специфичная терапия не является полностью персонализированной. Пары «донор — реципиент» легко генотипировать на отдельные МАГ или их панели [88]. Количество мутаций в лейкозах невелико, и неоантигены возникают значительно реже, чем в других типах опухолей [89], что делает МАГ особенно привлекательными именно для лечения этого типа злокачественных заболеваний.
МАГ имеют преимущество и перед поверхностными клеточными маркерами, как например CD19, являющимся маркером B-лимфоцитов и В-клеточных опухолей. CD19 — наиболее широко используемый антиген для Т-клеток с химерным антигенным рецептором (Chimeric Antigen Receptor T-cells, CAR-T) [90]. CAR-T-терапия показала высокую эффективность при лечении В-клеточных лейкозов, однако при ее проведении элиминируются в том числе здоровые В-клетки, что приводит к В-клеточной аплазии и увеличению риска инфекционных осложнений [91]. МАГ-специфичная терапия лишена этих ограничений, поскольку она селективно уничтожает клетки реципиента, не затрагивая клетки крови, происходящих из донорских гемопоэтических клеток. Несмотря на успехи в лечении CD19-положительных опухолей, CAR-T-терапия демонстрирует ограниченную применимость в лечении лейкозов миелоидного происхождения [92]. МАГ-специфичная терапия может быть универсальной для различных лейкозов. Однако МАГ как мишень для терапии опухолей имеет ограничения. Во-первых, МАГ-специфичная терапия может применяться только в контексте алло-ТГСК. Во-вторых, поскольку МАГ презентируются в конкретных аллелях HLA, МАГ-специфичную терапию можно использовать только у больных-носителей конкретного аллеля HLA. Обязательным условием является несовпадение по МАГ между донором и реципиентом. Для использования МАГ как терапевтической мишени необходимо, чтобы донор имел генотип МАГ–/–, а реципиент — МАГ+/+ или МАГ+/–. Максимальная доля иммуногенных несовпадений по конкретному МАГ составляет 25 %, что происходит при частоте иммуногенного аллеля в популяции 0,3 [62]. Лишь 10–15 % иммуногенных полиморфизмов имеют частоту, близкую к 0,3, и перспективны для клинического применения. Существует вероятность, что опухоль будет «ускользать» от иммунного ответа за счет выключения экспрессии гена, кодирующего МАГ или HLA, его презентирующего. Описано, что при трансплантации от HLA-несовместимого донора под действием клонального отбора клетки опухоли способны терять экспрессию HLA [93]. Аналогичный механизм может сделать их невидимыми и для МАГ-специфичных Т-клеток донора.
НА-1 — наиболее изученный минорный антиген. Он экспрессируется в здоровых гемопоэтических клетках и в клетках многих лейкозов, включая ОМЛ, миелодиспластические синдромы, Т- и В-клеточные ОЛЛ [87][94]. Пептид HA-1H (VLHDDLLEA) — продукт гена ARHGAP45, имеющего аденозиновый вариант однонуклеотидного полиморфизма (rs1801284, база данных dbSNP), презентирующийся на поверхности клетки в ассоциации с HLA-A*02:01 [36]. Индивиды с генотипом rs1801284 A/A или A/G имеют гистидиновый вариант (HA-1H, также называемый HA-1) и считаются HA-1-положительными. Напротив, HA-1-отрицательные лица с генотипом G/G имеют только аргининовый аллельный вариант (HA-1R, VLRDDLLEA), в связи с чем они могут иметь Т-клетки, специфичные к HA-1H. HA-1H и HA-1R образуются при расщеплении протеасомой белка ARHGAP45, оба пептида транспортируются в эндоплазматический ретикулум с помощью транспортера TAP [95]. Однако присутствие аргинина в третьей аминокислоте пептида значительно снижает аффинность связывания VLRDDLLEA с HLA-A*02:01 по сравнению с VLHDDLLEA [65], а HA-1H-специфические Т-клетки не распознают VLRDDLLEA при концентрациях пептида, которые в норме присутствуют на клетках. Следовательно, HA-1-специфические Т-клетки могут быть использованы после алло-ТГСК для избирательного уничтожения клеток лейкоза у HA-1-положительного больного без повреждения HA-1-отрицательных здоровых донорских гемопоэтических клеток. Примерно 50 % населения имеют HLA-A*02:01, а аллельные варианты HA-1 фенотипически сбалансированы в популяции (rs1801284 A/A 16 %, A/G 36 %, G/G 48 %). Таким образом, 52 % являются HA-1-положительными, 48 % — HA-1-отрицательными [87], а 25 % трансплантаций имеют иммуногенное несоответствие.
Остается открытым вопрос о связи минорного антигена НА-1 и РТПХ и, следовательно, безопасности, основанной на НА-1-терапии. Экспрессия источника пептида НА-1 — белка HMHA1 — практически отсутствует или происходит на очень низком уровне в негемопоэтических клетках [94][96]. В условиях in vitro HA-1-реактивные Т-клеточные клоны распознавали HA-1+ гемопоэтические клетки, но не распознавали негемопоэтические клетки с тем же генотипом [97]. Было показано отсутствие реактивности НА-1-специфичных клонов на образцы биопсии кожи из НА-1-позитивных доноров [98].
После алло-ТГСК HA-1-отрицательного донора HLA-A*02:01 и HA-1-положительному реципиенту HA-1-специфические Т-клетки были идентифицированы примерно у трети больных [50]. Сообщено о взаимосвязи между наличием HA-1-специфических Т-клеток и РТПХ, возникающей непосредственно после алло-ТГСК [99]. Однако HA-1-специфические T-клетки были идентифицированы также и у больных без РТПХ после алло-ТГСК с последующей инфузией донорских лимфоцитов [85][100].
Таким образом, в ряде работ иммунный ответ на НА-1 связывают с РТПХ [20][99][101–103]. В других исследованиях такой связи не находят [104–107]. Одним из возможных объяснений такого противоречия является то, что после алло-ТГСК гемопоэтические клетки реципиента в течение еще нескольких месяцев находятся в тканях до полного замещения гемопоэтическими клетками донора [87]. Для развития РТПХ CD8+ Т-лимфоцитам необходим прямой контакт с клетками-мишенями [108][109]. НА-1-специфичные Т-клоны, возникшие из наивных предшественников в условиях in vivo, могут мигрировать в ткани и атаковать гемопоэтические клетки реципиента, вызывая иммунный ответ на другие антигены [110]. Однако возникновение РТПХ менее вероятно при инфузии донорских лимфоцитов спустя несколько месяцев после алло-ТГСК [111] или при использовании генетически модифицированных лимфоцитов, имеющих рецептор с одной единственной специфичностью.
Уменьшение частоты развития РТПХ при отсрочке введения донорских лимфоцитов было показано и в экспериментах на мышах [112]. Уменьшенный риск возникновения РТПХ при отложенной инфузии донорских лимфоцитов объясняют также повышенным в первые месяцы после алло-ТГСК содержанием воспалительных цитокинов, облегчающим активацию МАГ-специфичных лимфоцитов донора [85]. У больных с РТПХ иммунный ответ направлен сразу на несколько МАГ, и нельзя выделить главенствующую роль НА-1-специфичных клеток в процессе РТПХ, что противоречит ранее показанной иммунодоминантности НА-1 [41].
В крови реципиентов, у которых не было РТПХ, были обнаружены НА-1-специфичные Т-лимфоциты, однако их клональное разнообразие было меньше, чем у больных с РТПХ. Таким образом, генетическое различие между донором и реципиентом по НА-1 само по себе не приводит к РТПХ. Поскольку пусковым механизмом РТПХ является воспалительный цитокиновый фон [113], то риск РТПХ можно уменьшить, если разнести по времени алло-ТГСК и трансфузию донорских лимфоцитов или использовать HA-1-специфичные генетически модифицированные лимфоциты.
Минорный антиген НА-2 был открыт в числе первых [41], тогда же было установлено, что он экспрессируется в гемопоэтической ткани, и отсутствует в клетках эпителия и эндотелия различных тканей [97]. Этот МАГ презентируется в HLA-A*02, имеет частоту иммуногенного аллеля в европейской популяции 95 % и является иммунодоминантным. Пептид НА-2 происходит из миозина I класса [114]. С помощью секвенирования генома установлено, что НА-2 — продукт гена MYO1G, имеющего два аллеля: иммуногенный MYO1GV (YIGEVLVSV) и неиммуногенный MYO1GM (YIGEVLVSM) [115]. Аминокислотная замена не приводит к значительному изменению аффинности связывания с HLA, а при добавлении экзогенного пептида Т-лимфоциты способны распознавать не только MYO1GV, но и MYO1GM. Однако исследование Т-клеточного ответа и масс-спектрометрический анализ связанных с HLA пептидов не показали способность эндогенного пептида MYO1GM презентироваться в HLA. Следовательно, причиной отсутствия иммуногенности может быть его слабое связывание с TAP или расщепление протеасомой на неподходящие для презентации в HLA фрагменты. Из-за большой частоты иммуногенного аллеля в европейской популяции иммуногенное несовпадение при трансплантации возникает редко, по сравнению с НА-1 имеется меньший объем информации о влиянии несовпадения по НА-2 на исход трансплантации. Анализ исходов трансплантации показывает меньшую вероятность рецидива лейкоза при несовпадении по НА-2 и большую вероятность РТПЛ [116]. При этом из-за ограниченной гемопоэтической тканью экспрессии, НА-2 не является причиной возникновения РТПХ [107], что подтверждается опытами с образцами кожи НА-2-позитивных доноров [98].
НА-2, как и НА-1, представляет собой перспективную мишень для посттрансплантационной иммунотерапии лейкозов. Инфузия донорских лимфоцитов при НА-2-несовместимой алло-ТГСК была ассоциирована с позитивным терапевтическим эффектом [100]. Был клонирован функциональный ТКР, специфичный к НА-2 [117], а затем был предложен метод терапии рецидивов лейкозов, основанный на внедрении в специфичные к цитомегаловирусу (ЦМВ) и вирусу Эпштейна — Барр (ВЭБ) Т-лимфоциты трансгенного НА-2-специфичного ТКР [118]. Установлена специфическая цитотоксичность модифицированных Т-лимфоцитов в отношении клеток лейкоза. В последующей работе была предпринята попытка увеличить эффективность терапии за счет увеличения аффинности НА-2-специфичного ТКР, что привело к возникновению кросс-реактивности на пептид SVGSVLLTV из гена CDH13, также презентируемого в HLA-A*02. Этот ген экспрессируется в здоровых фибробластах и кератиноцитах, что делает данный ТКР с увеличенной аффинностью непригодным для клинического применения [119]. В настоящее время испытания трансгенных НА-2-специфичных Т-лимфоцитов остаются на стадии in vitro, что связано с редкой частотой иммуногенного несовпадения при трансплантации в европейской популяции. Очень высокая частота MYO1GV в популяции приводит к тому, что лишь небольшая доля полностью HLA-совместимых пар «донор — реципиент» имеет иммунологическое несоответствие, дающее возможность использовать его в качестве мишени. По той же причине MYO1GV является почти универсальной мишенью для иммунотерапии при алло-ТГСК от гаплоидентичного донора, когда донор не имеет аллеля HLA-A*02:01, а реципиент имеет. В то же время в других популяциях аллельные варианты MYO1GV и MYO1GM имеют схожую частоту [120][121].
ACC1/2. Ген BCL2A1 принадлежит к семейству антиапоптотических генов Bcl-2. Два минорных антигена, ACC-1Y и ACC-2D, происходят из разных однонуклеотидных полиморфизмов в гене BCL2A1 и презентируются в аллельных вариантах молекул HLA-A*24:02 и HLA-B*44:03 соответственно [122]. BCL2A1 часто гиперэкспрессирован в злокачественных гемопоэтических клетках и является протоонкогеном. Потенциально опухоли будет труднее «уйти» от иммунного ответа за счет уменьшения экспрессии этого гена, что делает ACC-1 и ACC-2 перспективными мишенями для иммунотерапии [123][124]. Минорный антиген АСС-1 возникает благодаря полиморфизмам rs1138357 в первом экзоне гена BCL2A1, которые приводят к образованию двух пептидных вариантов — ACC-1Y (DYLQYVLQI) и ACC-1C (DYLQCVLQI), презентирующихся в HLA-A*24:02, оба из которых иммуногенны. Минорный антиген ACC-2 происходит из полиморфизма rs3826007, единственный иммуногенный пептид KEFEDDIINW презентируется в HLA-B*44:03. ACC-1Y-специфичные Т-клетки могут быть получены от доноров с аллелем HLA-A*24:02, гомозиготных по полиморфизму (rs1138357, GG, кодирующему DYLQCVLQI). Точно так же ACC-2-специфические Т-клетки могут быть получены доноров с аллелем HLA-B*44:03, гомозиготных по полиморфизму (rs3826007, GG, кодирующему KEFEDGIINW). Нами ранее был клонирован ТКР, специфичный к минорному антигену ACC-1Y, подтверждена его функциональность и отсутствие распознавания альтернативного аллеля [125]. Для антигена ACC-2 последовательности распознающих его ТКР на сегодняшний момент не были опубликованы.
ACC-1 и ACC-2 являются перспективными мишенями для Т-клеточной иммунотерапии. На основании распространенности аллелей HLA-A*24:02 и HLA-B*44:03 и частоты распространения иммуногенных и неиммуногенных вариантов ACC-1 и ACC-2, частота иммуногенного несовпадения при трансплантации от родственных и неродственных доноров составляет 2,8 и 5,2 % для ACC-1 и 3,6 и 6,7 % для ACC-2 соответственно [126].
Существуют разногласия относительно того, насколько избирательно BCL2A1 экспрессируется в гемопоэтических клетках. Анализ экспрессии генов с помощью Нозерн-блоттинга [122], количественной полимеразной цепной реакции и РНК-микрочипов [127] показывает экспрессию исключительно в гемопоэтических клетках. Однако показано [128], что экспрессия BCL2A1 может быть увеличена в негемопоэтических клетках (мезенхимных стромальных клетках) за счет одновременного воздействия интерферона-гамма и фактора некроза опухоли, что впоследствии было подтверждено другим исследованием [127]. Впрочем, клиническая значимость этого обнаруженного in vitro феномена не подтверждается. В двух исследованиях [127][128] негемопоэтические клетки, имеющие аллели ACC-1Y и ACC-2D, не распознавались Т-клеточными клонами без стимуляции экзогенными цитокинами. Более того, анализ результатов ТГСК у 320 больных, имевших аллель HLA-A*24:02, не показал увеличения частоты РТПХ при несовпадении по ACC-1 [129]. Из этих данных следует, что иммунотерапия с использованием ACC-1Y не несет риска развития РТПХ.
Было показано, что ACC-1-и ACC-2-специфичные Т-клетки in vitro обладают способностью лизировать первичные лейкозные клетки, полученные от реципиентов гемопоэтических стволовых клеток [122]. В последующем исследовании с помощью окраски HLA-тетрамером ACC-1-специфичные Т-лимфоциты были выявлены в костном мозге и периферической крови у больного спустя 14 мес. после трансплантации от HLA-совместимого ACC-1-отрицательного донора [130]. ACC-1-специфические Т-клетки, выделенные из костного мозга, пролиферировали в ответ на стимуляцию пептидом ACC1 и проявляли цитотоксичность в отношении клеточной линии, представляющей эндогенный пептид ACC-1, демонстрируя способность ACC-1-специфичных Т-лимфоцитов формировать иммунологическую память, способствующую защите от посттрансплантационного рецидива. Тем не менее, BCL2A1 экспрессируется на высоком уровне не при всех лейкозах. Хотя цитотоксические Т-клетки, имеющие высокоаффинный ТКР, способны уничтожать клетки с низким уровнем экспрессии мишени, перед клиническим применением ACC-специфичной терапии целесообразно оценивать экспрессию этого гена и проводить цитотоксические тесты на бластных клетках конкретных больных.
Наиболее простой вариант посттрансплантационной иммунотерапии заключается в трансфузии реципиенту лимфоцитов донора. Эта процедура направлена не только на профилактику или терапию рецидива лейкоза, но и на терапию оппортунистических инфекций, риск возникновения которых сильно повышен из-за связанного с ТГСК кондиционирования и вызываемой им лимфопении [131].
Наибольшее количество специфичных к минорным антигенам реципиента Т-клеточных клонов содержится среди наивных клеток. При контакте с антигеном они способны активироваться и вызывать иммунный ответ на опухолевые клетки при лейкозе [21]. В экспериментальной модели на мышах [132] адоптивный перенос CD8+ лимфоцитов, полученных из мышей, иммунизированных клетками, несущими МАГ, привел к излечению мышей от лейкоза без возникновения РТПХ.
В клиническом исследовании [100] трем HA-1-и/или НА-2-позитивным больным с рецидивом лейкоза производили инфузию лимфоцитов от HA-1-и/или НА-2-отрицательных доноров. У всех 3 больных были достигнуты ремиссия лейкоза и 100 % донорский химеризм. Экспансию НА-1/2-специфичных клонов в крови реципиентов подтверждали окраской HLA-тетрамерами, направленная цитотоксичность этих клонов была подтверждена in vitro [133].
Количество наивных лимфоцитов, специфичных к МАГ, непосредственно в донорской крови невелико: для НА-1 частота наивных клонов оценивается как 2,47 × 10–6 [134]. В клинических исследованиях метод трансфузии донорских Т-лимфоцитов, полученных с помощью ex vivo экспансий, специфичных к бластным клеткам больных лейкозами, показал умеренную эффективность [135]. Конкретные антигены оставались неизвестными, однако предполагают, что в основном клеточные экспансии были получены на МАГ.
В исследовании [136] 7 больным с рецидивом В-клеточного ОЛЛ или миелодиспластическим синдромом были перелиты Т-лимфоциты, специфичные к МАГ, полученные в результате ex vivo экспансий. Периферические мононуклеары от больных после трансплантации, содержавшие клетки донорского происхождения, стимулировали 3 раза облученными мононуклеарами этих же больных, но до трансплантации. Отбирали CD8+ Т-клеточные клоны, которые обладали селективной цитотоксичностью в отношении В-лимфобластных клеток реципиентского, но не донорского происхождения. Отобранные таким образом клоны характеризовали по уникально перестроенной бета-цепи ТКР, проводили экспансию и вводили больным 3–9 × 10 9 клеток. У 5 из 7 больных была достигнута временная ремиссия, у 2 из них наблюдалась умеренная РТПХ с поражением легких. Для 3 больных с помощью секвенирования генома были определены гены, на полиморфные пептиды которых были получены цитотоксические клоны: P2RX7, DPH1, DDX3Y. Это исследование подтверждает возможность использования МАГ в качестве мишеней для иммунотерапии лейкозов.
В исследовании [137] получали клеточный продукт, содержащий Т-лимфоциты, специфичные к НА-1, методом антиген-специфичной экспансии, и вводили 3 больным. Ни у одного из больных не было РТПХ, однако и клинического ответа не наблюдалось. Недостаточная эффективность метода в обоих исследованиях связана с неоптимальными условиями культивирования, которые привели к истощению культуры и ограниченному времени циркуляции лимфоцитов после трансфузии. В дальнейшем были предложены методы оптимизации условий in vitro экспансии антиген-специфичных клеток: культивирование с интерлейкином-7 и интерлейкином-15 [138] или блокада сигнального пути Akt/протеинкиназы B в лимфоцитах во время экспансии [139].
Естественный МАГ-специфичный иммунный ответ можно усилить за счет введения дендритно-клеточной вакцины параллельно с инфузией донорских лимфоцитов [140]. Девяти больным множественной миеломой (ММ) или с рецидивом ММ после алло-ТГСК вводили донорские лимфоциты, а также донорские дендритные клетки моноцитарного происхождения, презентирующие экзогенные пептиды МАГ. Пары «донор — реципиент» имели несовпадение по минорным антигенам HA-1 и/или UTA2–1 и/или LRH-1. Ни у одного из 5 больных не было РТПХ, у 5 больных МАГ-специфичные лимфоциты детектировались в течение 20 недель. У этих больных прогрессию болезни удалось остановить на 3,5–10 мес.
Дендритно-клеточную вакцину исследовали также в работе [141]. Для 4 из 15 больных, не ответивших на первичную инфузию лимфоцитов, была получена вакцина, презентирующая МАГ НА-1, НА-2 или ACC. У 1 из 4 больных достигнута ремиссия. Хотя за длительное время испытания дендритно-клеточные вакцины показали свою безопасность, терапевтический успех оценивается как неоднозначный [142].
Альтернативный и считающийся в настоящее время более перспективным подход заключается в получении Т-лимфоцитов, распознающих МАГ за счет их генетической модификации, например переносе МАГ-специфичного Т-клеточного рецептора в донорские лимфоциты с помощью вирусного вектора.
В одном из исследований был предложен оригинальный подход, заключающийся в использовании для модификации трансгенным ТКР донорских Т-лимфоцитов, специфичных к ЦМВ или ВЭБ. Предполагаемыми преимуществами такого подхода являлось, во-первых, то, что противовирусная специфичность Т-лимфоцитов должна была способствовать их размножению in vivo и одновременно осуществлять функцию профилактики ЦМВ-инфекции после трансплантации; во-вторых, вирусная специфичность ТКР должна исключить аллореактивность Т-лимфоцитов и потенциальное развитие РТПХ [143]. Модифицированные специфичным к НА-1 ТКР лимфоциты демонстрировали направленную цитотоксическую активность in vitro в отношении лимфобластной клеточной линии, несущей экзогенный пептид HA-1, а также бластных клеток, полученных от больных [144].
Этот подход был применен в первой фазе клинического исследования [145], в котором приняли участие 9 НА-1-позитивных больных с рецидивом ОМЛ после алло-ТГСК. У 4 из них доноры были ЦМВ-отрицательные, поэтому для них клеточный продукт изготовить было невозможно. Состав клеточного продукта сильно различался между больными: вводили от 3 до 283 × 10 6 клеток, из них Т-клеток было 96–99 %, вирус-специфичными были 74–100 %, а модифицированными трансгенным ТКР были 11–41 %. По результатам исследования, из 5 больных, которым вводили клеточный продукт, 1 умер от рецидива ОМЛ, 1 — от посттрансплантационного лимфопролиферативного синдрома и 1 — от бактериальной и грибковой инфекций, выжили 2 больных. Ни у одного не было признаков РТПХ. В группе, в которой не вводили клеточный продукт, 1 больной умер от бактериальной инфекции. По результатам этих исследований можно сделать вывод о низкой эффективности и ограниченной применимости подхода с использованием вирус-специфичных лимфоцитов как модифицируемой популяции. Трансгенные лимфоциты демонстрировали функциональный ответ на бластные клетки больных ОМЛ, полученные в момент рецидива, следовательно, причина неэффективности заключалась не в потере экспрессии МАГ клетками лейкоза. Персистенция модифицированных клеток наблюдалась у 3 из 5 больных, причем эти больные умерли впоследствии. Причина неудачи объясняется недостаточным количеством модифицированных лимфоцитов. Вирус-специфичные лимфоциты, использованные для модификации, принадлежат к популяциям клеток памяти и эффекторам, обладающим меньшим пролиферативным потенциалом по сравнению с наивными клетками [146]. Распознавание лимфоцитами вирусных антигенов дает достаточный стимул для поддержания трансгенной популяции Т-клеток. Однако из-за оптимизации кодонов экспрессия трансгенного ТКР подавила экспрессию эндогенного вирус-специфичного ТКР. Этими факторами объясняется плохая персистенция лимфоцитов в организме реципиентов. Этот метод невозможно стандартизировать и масштабировать из-за малого количества стартового материала для модификации: количество вирус-специфичных клеток оценивается примерно в 1 % от общего пула Т-лимфоцитов [147].
В работе [148] выбран другой подход к терапии: НА-1-специфичные лимфоциты были получены путем трансдукции тотальной фракции CD8+ и CD4+ клеток. В лентивирусный вектор помимо ТКР включены селективный маркер (укороченный CD34), корецептор CD8 и индуцибельная каспаза iCasp9. Использование каспазы позволяет уничтожить трансгенные лимфоциты в случае их неконтролируемой экспансии или развития РТПХ за счет введения AP1903 (Rimiducid), приводящего к димеризации iCasp9 и запуску апоптоза. В настоящее время проводится набор больных для I фазы клинического исследования (NCT03326921), которое должно завершиться в 2024 г.
Количество клинических исследований терапии на основе МАГ очень невелико (табл.) по сравнению с испытаниями CAR-T, количество которых превышает 500 [90]. Также мало число больных, принимающих участие в каждом исследовании. Это объясняется трудностью поиска пары «донор — реципиент», обладающей нужным несовпадением по МАГ, для НА-1 это приблизительно 25 % от общего числа HLA-A*02-совместимых трансплантаций [149].
Таблица. Клинические исследования с использованием МАГ
Table. Clinical trials with Minor Histocompatibility Antigens
|
Публикация/номер клинического исследования Publication/clinical trial number |
Антигены Antigens |
Метод получения клеточного продукта Cell product manufacturing method |
Число больных Number of patients |
Результат Result |
Диагноз пациентов Patient diagnosis |
|
[100] |
HA-1, HA-2, Y-хромосомные антигены |
Инфузия донорских лимфоцитов Donor lymphocyte infusion |
3 |
Ремиссия Remission |
Рецидив ОМЛ Relapsed AML |
|
NCT00107354 [136] |
P2RX7, DPH1, DDX3Y |
Экспансия донорских Т-лимфоцитов ex vivo Donor T-lymphocyte ex vivo expansion |
7 |
Транзиторная ремиссия у 5 5 patients had transient remission |
Рецидив ОМЛ Relapsed AML |
|
[137] |
НА-1 |
Экспансия донорских Т-лимфоцитов ex vivo Donor T-lymphocyte ex vivo expansion |
3 |
Нет клинического ответа No clinical response |
Рецидив ОМЛ Relapsed AML |
|
[140] |
LRH-1, UTA2–1, HA-1 |
Инфузия донорских лимфоцитов и пептидная дендритно-клеточная вакцина Donor lymphocyte infusion and peptide-pulsed dendritic cell vaccine |
9 |
У 5 больных детектировались МАГ-специфичные лимфоциты и замедлилось развитие болезни MiHA-specific CD8 T-cells were found in 5 patients and disease progression slowed |
Рецидив ММ и персистирующая ММ Relapsed or recurrent ММ |
|
[141] |
HA-1, HA-2, ACC1 |
Инфузия донорских лимфоцитов и пептидная дендритно-клеточная вакцина Donor lymphocyte infusion and peptide-pulsed dendritic cell vaccine |
15 |
Ремиссия у 1 больного, хроническое заболевание/прогрессия — у остальных 1 patient had complete remission, the rest had chronic disease/progression of the disease |
ММ |
|
NCT04464889 [145] |
HA-1 |
Трансдукция вирус-специфичных лимфоцитов Virus-specific lymphocytes transduction |
9 больных участвовали в первой фазе, 20 больных планируется набрать во второй фазе 9 in phase I, 20 expected in phase II |
Нет клинического ответа, смерть одного больного от рецидива ОМЛ No clinical response, death of one patient from AML |
Рецидив ОМЛ Relapsed AML |
|
NCT03326921 [148] |
HA-1 |
Трансдукция донорских CD4 и CD8 лимфоцитов Donor CD4 and CD8 T-cell transduction |
Набор пациентов Patient recruitment |
– |
– |
|
NCT03091933 |
Не разглашаются Not disclosed |
Экспансия донорских Т-лимфоцитов ex vivo Donor T-lymphocyte ex vivo expansion |
20 |
Умеренный клинический ответ: остановка прогрессии заболевания у части больных, признаки РТПХ Disease progression stopped in few patients, GVHD symptoms |
Рецидив ОЛЛ, ОМЛ, ХЛЛ, НХЛ, ММ, ХЛ, рецидивирующие миелодиспластические синдромы Relapsed ALL, AML, CLL, NHL, HL, MM, relapsed myelodysplastic syndromes |
|
2018–002752–33 |
UTA2–1 |
мРНК дендритно-клеточная вакцина с сайленсингом PD1-L РНК-интерференцией mRNA dendritic сell vaccine with PD-1 siRNA silencing |
Набор больных Patient recruitment |
– |
ММ, НХЛ, ХЛЛ, ОМЛ MM, NHL, CLL, AML |
|
2012–002435–28 |
Не разглашаются Not disclosed |
мРНК дендритно-клеточная вакцина с сайленсингом PD1-L РНК-интерференцией mRNA dendritic сell vaccine with PD-1 siRNA silencing |
Набор больных Patient recruitment |
– |
НХЛ, ММ, ХЛЛ, ХМЛ, ОМЛ, миелодисплазия NHL, MM, NHL, MM, CLL, CML, AML, myelodysplasia |
|
NCT02528682 |
HA-1, LRH-1, ARHGDIB |
мРНК дендритно-клеточная вакцина с сайленсингом PD1-L РНК-интерференцией mRNA dendritic сell vaccine with PD-1 siRNA silencing |
10 |
Испытания завершены, результаты еще не опубликованы Trials finished, no results published yet |
ОМЛ, миелодисплазия, ОЛЛ, ХМЛ, ХЛЛ, ММ, НХЛ AML, myelodysplasia, ALL, CML, CLL, MM, NHL |
При внедрении трансгенного ТКР возможно формирование гетеродимеров между нативными и трансгенными альфа- и бета-цепями ТКР. Это может приводить к образованию новых ТКР, обладающих неизвестной специфичностью, и потенциально вызвать РТПХ [150]. Нокаут эндогенного ТКР системой CRISPR/Cas9 позволяет избежать возникновения новой специфичности и улучшить эффекторные качества клеток за счет устранения внутриклеточной конкуренции между разными ТКР [151] (рис. 2).
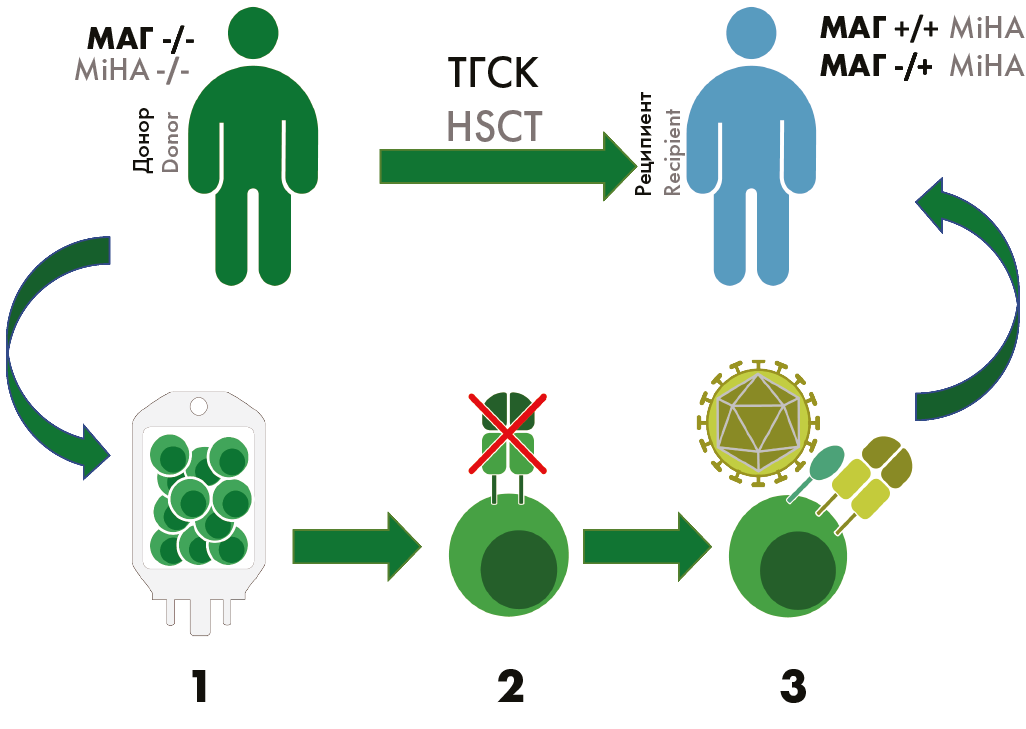
Рисунок 2. Схема МАГ-специфичной терапии лейкозов, совмещенной с ТГСК. 1) От донора, не имеющего МАГ, выделяют и активируют Т-лимфоциты. 2) С помощью CRISPR/Cas проводят нокаут эндогенного ТКР. 3) Лимфоциты, содержащие МАГ-специфичный трансгенный ТКР, доставленный с помощью лентивирусного вектора, вводят больному
Figure 2. Plan of MiHA-specific leukemia immunotherapy combined with HSCT. 1) Isolation and activation of T-cells from a MiHA-negative donor. 2) CRISPR/Cas mediated knockout of endogenous TCR 3) Lymphocytes with a MiHA-specific transgenic TCR modified by lentiviral vector are transfused to a patient
Как альтернатива трансгенному ТКР был разработан химерный рецептор, специфичный к НА-1 [152]. Использование такого рецептора будет иметь те же ограничения, связанные с подбором пары «донор — реципиент», но потенциально химерный рецептор, обладающий большей аффинностью, сможет распознавать антиген даже при низком уровне его экспрессии.
Таким образом, терапия или профилактика рецидивов злокачественных заболеваний системы крови после алло-ТГСК, направленная на МАГ, является перспективным методом лечения за счет возможности индукции РТПЛ в отсутствии РПТХ. МАГ имеют ряд существенных преимуществ перед другими антигенами в качестве мишеней иммунотерапии. Отсутствие на сегодняшний день значительных успехов в области МАГ-специфичной терапии в большей степени связано с несовершенством методик получения клеточного продукта, использованного в предшествующих испытаниях. Применение современных протоколов культивации Т-лимфоцитов, а также использование для модификации популяций, обладающих большим пролиферативным потенциалом, таких как наивные [153] и стволовые клетки памяти [154], должны повысить эффективность терапии. Учитывая опыт неудачных клинических испытаний и опыт, полученный при использовании CAR-T-терапии, необходимо модифицировать процесс получения Т-лимфоцитов, чтобы повысить их долгосрочную выживаемость и функциональную активность.
Наиболее перспективным подходом представляется не выращивание антиген-специфичных экспансий, а получение антиген-специфичных Т-лимфоцитов за счет внедрения гена Т-клеточного рецептора с известной специфичностью. Это позволит получать стандартизированные Т-лимфоциты с заранее известными свойствами и изученной кросс-реактивностью. Принципиальные ограничения подхода, связанные с небольшой долей больных, у которых возможно таргетировать конкретный МАГ, могут быть преодолены за счет применения МАГ-специфичной терапии в контексте алло-ТГСК от гаплоидентичного донора. По-прежнему остается актуальным поиск новых МАГ, преимущественно экспрессированных в клетках гемопоэтической системы и с повышенной экспрессией в опухолевых клетках. Особенно актуальны МАГ, происходящие из функционально значимых для опухоли генов. Информация, накопленная в предшествующих клинических исследованиях, и арсенал подходов генетического редактирования и производства клеточного продукта, отработанный в области CAR-T-терапии, создали предпосылки к тому, чтобы в ближайшем будущем стало возможным создание высокоспецифичной и эффективной терапии лейкозов, направленной на МАГ.
1. Савченко В.Г., Любимова Л.С., Паровичникова Е.Н., и др. Трансплантации аллогенных и аутологичных гемопоэтических стволовых клеток при острых лейкозах (итоги 20-летнего опыта). Терапевтический архив. 2007; 79(7):30–5.
2. Giebel S., Labopin M., Potter M., et al. Comparable results of autologous and allogeneic haematopoietic stem cell transplantation for adults with Philadelphiapositive acute lymphoblastic leukaemia in fi rst complete molecular remission: An analysis by the Acute Leukemia Working Party of the EBMT. Eur J Cancer. 2018; 96: 73–81. DOI: 10.1016/j.ejca.2018.03.018.
3. Schmid C., de Wreede L.C., van Biezen A., et al. Outcome after relapse of myelodysplastic syndrome and secondary acute myeloid leukemia following allogeneic stem cell transplantation: A retrospective registry analysis on 698 patients by the Chronic Malignancies Working Party of the European Society of Blood and Marrow Transplantation. Haematologica. 2018; 103(2): 237–45. DOI: 10.3324/haematol.2017.168716.
4. Rautenberg C., Germing U., Haas R., et al. Relapse of acute myeloid leukemia after allogeneic stem cell transplantation: Prevention, detection, and treatment. Int J Mol Sci. 2019; 20(1): 228. DOI: 10.3390/ijms20010228.
5. Schmid C., Labopin M., Nagler A., et al. Treatment, risk factors, and outcome of adults with relapsed AML after reduced intensity conditioning for allogeneic stem cell transplantation. Blood. 2012; 119(6): 1599–606. DOI: 10.1182/blood-2011-08-375840.
6. McDonald G.B., Sandmaier B.M., Mielcarek M., et al. Survival, nonrelapse mortality, and relapse-related mortality after allogeneic hematopoietic cell transplantation: Comparing 2003–2007 versus 2013–2017 cohorts. Ann Intern Med. 2020; 172(4): 229–39. DOI: 10.7326/m19-2936.
7. Kröger N. Hematopoietic stem cell transplantation and cellular therapies. In: Carreras E., Dufour C., Mohty M., Kröger N. (eds) The EBMT Handbook. Springer, Cham. https://doi.org/10.1007/978-3-030-02278-5_58.
8. Kirtonia A., Pandya G., Sethi G., et al. A comprehensive review of genetic alterations and molecular targeted therapies for the implementation of personalized medicine in acute myeloid leukemia. J Mol Med (Berl). 2020; 98(8): 1069–91.DOI: 10.1007/s00109-020-01944-5.
9. Савченко В.Г., Менделеева Л.П., Паровичникова Е.Н., и др. Способ лечения рецидива острого миелоидного лейкоза после трансплантации аллогенных гемопоэтических стволовых клеток. Патент № RU 2538799 C1, 2019.
10. Butturini A., Bortin M.M., Gale R.P. Graft-versus-leukemia following bone marrow transplantation. Bone Marrow Transpl. 1987; 2(3): 233–42.
11. Weiden P.L., Flournoy N., Thomas E.D., et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med. 1979; 300(19): 1068–73. DOI: 10.1056/nejm197905103001902.
12. Jones R., Ambinder R., Piantadosi S., Santos G. Evidence of a graft-versuslymphoma effect associated with allogeneic bone marrow transplantation. Blood. 1991; 77(3): 649–53. DOI: 10.1182/blood.V77.3.649.649.
13. Thomas E.D. A history of haemopoietic cell transplantation. Br J Haematol. 1999; 105(2): 330–9. DOI: 10.1111/j.1365-2141.1999.01337.x.
14. Slavin R.E., Woodruff J.M. The pathology of bone marrow transplantation. Pathol Annu. 1974; 9(0): 291–344.
15. Ефимов Г.А., Вдовин А.С., Григорьев А.А. и др. Иммунобиология острой реакции «трансплантат против хозяина». Медицинская иммунология. 2015; 17(6): 499–516. DOI: 10.15789/1563-0625-2015-6-499-516.
16. Lakkis F.G., Lechler R.I. Origin and biology of the allogeneic response. Cold Spring Harb Perspect Med. 2013; 3(8): a014993. DOI: 10.1101/cshperspect.a014993.
17. D’Orsogna L., Nguyen T., Claas F., et al. Endogenous peptide dependent alloreactivity: New scientifi c insights and clinical implications. Tissue Antigens. 2013; 81(6): 399–407. DOI: 10.1111/tan.12115.
18. Korngold R., Sprent J. Features of T cells causing H-2-restricted lethal graftvs.-host disease across minor histocompatibility barriers. J Exp Medicine. 1982; 155(3): 872–83. DOI: 10.1084/jem.155.3.872.
19. Korngold R, Sprent J. Lethal GVHD across minor histocompatibility barriers: Nature of the effector cells and role of the H 2 complex. Immunol Rev. 1983; 71:5–30. DOI: 10.1111/j.1600-065x.1983.tb01066.x.
20. Goulmy E., Schipper R., Pool J., et al. Mismatches of minor histocompatibility antigens between HLA-identical donors and recipients and the development of graft-versus-host disease after bone marrow transplantation. N Engl J Med. 1996; 334(5): 281–5. DOI: 10.1056/NEJM199602013340501.
21. Bleakley M., Otterud B.E., Richardt J.L., et al. Leukemia-associated minor histocompatibility antigen discovery using T-cell clones isolated by in vitro stimulation of naive CD8+ T cells. Blood. 2010; 115(23): 4923–33. DOI: 10.1182/blood-2009-12-260539.
22. Weiden P.L., Sullivan K.M., Flournoy N., et al. Antileukemic effect of chronic graft-versus-host disease — contribution to improved survival after allogeneic marrow transplantation. N Engl J Med. 1981; 304(25): 1529–33. DOI: 10.1056/nejm198106183042507.
23. Horowitz M.M., Gale R.P., Sondel P.M., et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990; 75(3): 555–62. DOI: 10.1182/blood.V75.3.555.555.
24. Inamoto Y., Flowers M.E.D., Lee S.J., et al. Infl uence of immunosuppressive treatment on risk of recurrent malignancy after allogeneic hematopoietic cell transplantation. Blood. 2011; 118(2): 456–63. DOI: 10.1182/blood-2011-01-330217.
25. Kersey J.H., Weisdorf D., Nesbit M.E., et al. Comparison of autologous and allogeneic bone marrow transplantation for treatment of high-risk refractory acute lymphoblastic leukemia. N Engl J Med. 1987; 317(8): 461–7. DOI: 10.1056/nejm198708203170801.
26. Gratwohl A., Sureda A., Cornelissen J., et al. Alloreactivity: The Janus-face of hematopoietic stem cell transplantation. Leukemia. 2017; 31(8): 1752–9. DOI: 10.1038/leu.2017.79.
27. Falkenburg J.H.F., Marijt W.A.F., Heemskerk M.H.M., Willemze R. Minor histocompatibility antigens as targets of graft-versus-leukemia reactions. Curr Opin Hematol. 2002; 9(6): 497–502. DOI: 10.1097/00062752-200211000-00005.
28. Bevan M.J. The major histocompatibility complex determines susceptibility to cytotoxic T cells directed against minor histocompatibility antigens. J Exp Medicine. 1975; 142(6): 1349–64. DOI: 10.1084/jem.142.6.1349.
29. Korngold R., Sprent J. Lethal graft-versus-host disease after bone marrow transplantation across minor histocompatibility barriers in mice. Prevention by removing mature T cells from marrow. J Exp Medicine. 1978; 148(6): 1687–98. DOI: 10.1084/jem.148.6.1687.
30. Hamilton B.L., Bevan M.J., Parkman R. Anti-recipient cytotoxic T lymphocyte precursors are present in the spleens of mice with acute graft versus host disease due to minor histocompatibility a e foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature. 1987; 329(6139): 512–8. DOI: 10.1038/329512a0.
31. Wallny H-J., Rammensee H-G. Identifi cation of classical minor histocompatibility antigen as cell-derived peptide. Nature. 1990; 343(6255): 275–8. DOI: 10.1038/343275a0.
32. Goulmy E. Human minor histocompatibility antigens: New concepts for marrow transplantation and adoptive immunotherapy. Immunol Rev. 1997; 157: 125–40. DOI: 10.1111/j.1600-065x.1997.tb00978.x.
33. Kernan N.A., Bartsch G., Ash R.C., et al. Analysis of 462 transplantations from unrelated donors facilitated by the National Marrow Donor Program. New Engl J Med. 1993; 328(9): 593–602. DOI: 10.1056/nejm199303043280901.
34. Kernan N., Dupont B. Minor histocompatibility antigens and marrow transplantation. N Engl J Med. 1996; 334(5): 323–4. DOI: 10.1056/NEJM199602013340510.
35. den Haan J., Meadows L., Wang W., et al. The minor histocompatibility antigen HA-1: A diallelic gene with a single amino acid polymorphism. Science. 1998; 279(5353): 1054–7. DOI: 10.1126/science.279.5353.1054.
36. von Boehmer H., Hafen K. Minor but not major histocompatibility antigens of thymus epithelium tolerize precursors of cytolytic T cells. Nature. 1986; 320(6063): 626–8. DOI: 10.1038/320626a0.
37. Vincent K., Roy D-C., Perreault C. Next-generation leukemia immunotherapy. Blood. 2011; 118(11): 2951–9. DOI: 10.1182/blood-2011-04-350868.
38. Goulmy E., Gratama J.W., Blokland E., et al. A minor transplantation antigen detected by MHC-restricted cytotoxic T lymphocytes during graft-versus-host disease. Nature. 1983; 302(5904): 159–61. DOI: 10.1038/302159a0.
39. Rötzschke O., Falk K., Wallny H., et al. Characterization of naturally occurring minor histocompatibility peptides including H-4 and H-Y. Science. 1990; 249(4966): 283–7. DOI: 10.1126/science.1695760.
40. van Els C., D’Amaro J., Pool J., et al. Immunogenetics of human minor histocompatibility antigens: Their polymorphism and immunodominance. Immunogenetics. 1992; 35(3): 161–5. DOI: 10.1007/BF00185109.
41. Rufer N., Wolpert E., Helg C., et al. HA-1 and the SMCY-derived peptide FIDSYICQV (H-Y) are immunodominant minor histocompatibility antigens after bone marrow transplantation. Transplantation. 1998; 66(7): 910–6. DOI: 10.1097/00007890-199810150-00016.
42. Korngold R., Leighton C., Mobraaten L.E., Berger M.A. Inter-strain graft-vs.-host disease T-cell responses to immunodominant minor histocompatibility antigens. Biol Blood Marrow Transplant. 1997; 3(2): 57–64.
43. Pion S., Fontaine P., Baron C., et al. Immunodominant minor histocompatibility antigens expressed by mouse leukemic cells can serve as effective targets for T cell immunotherapy. J Clin Invest. 1995; 95(4): 1561–8. DOI: 10.1172/jci117829.
44. Yewdell J.W., Bennink J.R. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Ann Rev Immunol. 1999; 17: 51–88. DOI: 10.1146/annurev.immunol.17.1.51.
45. Wettstein P.J. Immunodominance in the T-cell response to multiple non-H-2 histocompatibility antigens. II. Observation of a hierarchy among dominant antigens. Immunogenetics. 1986; 24(1): 24–31. DOI: 10.1007/bf00372294.
46. Chen W., Khilko S., Fecondo J., et al. Deteromplex class I-restricted antigenic peptides is explained by class Ipeptide affi nity and is strongly infl uenced by nondominant anchor residues. J Exp Medicine. 1994; 180(4): 1471–83. DOI: 10.1084/jminant selection of major histocompatibility cem.180.4.1471.
47. Pion S., Fontaine P., Desaulniers M., et al. On the mechanisms of immunodominance in cytotoxic T lymphocyte responses to minor histocompatibility antigens. Eur J Immunol. 1997; 27(2): 421–30. DOI: 10.1002/eji.1830270212.
48. Mori S., El-Baki H., Mullen C. Analysis of immunodominance among minor histocompatibility antigens in allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2003; 31(10): 865–75. DOI: 10.1038/sj.bmt.1704021.
49. Hobo W., Broen K., van der Velden W., et al. Association of disparities in known minor histocompatibility antigens with relapse-free survival and graftversus-host disease after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2013; 19(2): 274–82. DOI: 10.1016/j.bbmt.2012.09.008.
50. Korngold R., Wettstein P.J. Immunodominance in the graft-vs-host disease T cell response to minor histocompatibility antigens. J Immunol. 1990; 145(12): 4079–88.
51. Goulmy E., Termijtelen A., Bradley B.A., Rood J.J.V. Y-antigen killing by T cells of women is restricted by HLA. Nature. 1977; 266(5602): 544–5. DOI: 10.1038/266544a0.
52. Antón L.C., Yewdell J.W. Translating DRiPs: MHC class I immunosurveillance of pathogens and tumors. J Leukoc Biol. 2014; 95(4): 551–62. DOI: 10.1189/jlb.1113599.
53. Apcher S., Millot G., Daskalogianni C., et al. Translation of pre-spliced RNAs in the nuclear compartment generates peptides for the MHC class I pathway. Proc Natl Acad Sci U S A. 2013; 110(44): 17951–6. DOI: 10.1073/pnas.1309956110.
54. Granados D., Yahyaoui W., Laumont C.M., et al. MHC I-associated peptides preferentially derive from transcripts bearing miRNA response elements. Blood. 2012; 119(26): e181–91. DOI: 10.1182/blood-2012-02-412593.
55. Laumont C.M., Daouda T., Laverdure J-P., et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat Commun. 2016; 7: 10238. DOI: 10.1038/ncomms10238.
56. Hanna J., Guerra-Moreno A., Ang J., Micoogullari Y. Protein degradation and the pathologic basis of disease. Am J Pathology. 2018; 189(1): 94–103. DOI: 10.1016/j.ajpath.2018.09.004.
57. Kisselev A.F., Akopian T.N., Woo K.M., Goldberg A.L. The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes implications for understanding the degradative mechanism and antigen presentation. J Biol Chem. 1999; 274(6): 3363–71. DOI: 10.1074/jbc.274.6.3363.
58. Pearson H., Daouda T., Granados D., et al. MHC class I-associated peptides derive from selective regions of the human genome. J Clin Invest. 2016; 126(12): 4690–701. DOI: 10.1172/JCI88590.
59. Abele R., Tampé R. Function of the transport complex TAP in cellular immune recognition. Biochim Biophys Acta. 1999; 1461(2): 405–19. DOI: 10.1016/s0005-2736(99)00171-6.
60. Rock K.L., Goldberg A.L. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu Rev Immunol. 1999; 17: 739–79. DOI: 10.1146/annurev.immunol.17.1.739.
61. Bykova N.A., Malko D.B., Efi mov G.A. In silico analysis of the minor histocompatibility antigen landscape based on the 1000 Genomes Project. Front Immunol. 2018; 9: 1819. DOI: 10.3389/fi mmu.2018.01819.
62. Spierings E., Brickner A.G., Caldwell J.A., et al. The minor histocompatibility antigen HA-3 arises from differential proteasome-mediated cleavage of the lymphoid blast crisis (Lbc) oncoprotein. Blood. 2003; 102(2): 621–9. DOI: 10.1182/blood-2003-01-0260.
63. Brickner A.G., Warren E.H., Caldwell J.A., et al. The immunogenicity of a new human minor histocompatibility antigen results from differential antigen processing. J Exp Med. 2001; 193(2): 195–206. DOI: 10.1084/jem.193.2.195.
64. Spierings E., Gras S., Reiser J-B., et al. Steric hindrance and fast dissociation explain the lack of immunogenicity of the minor histocompatibility HA-1Arg Null allele. J Immunol. 2009; 182(8): 4809–16. DOI: 10.4049/jimmunol.0803911.
65. de Rijke B., van Horssen-Zoetbrood A., Beekman J.M., et al. A frameshift polymorphism in P2X5 elicits an allogeneic cytotoxic T lymphocyte response associated with remission of chronic myeloid leukemia. J Clin Invest. 2005; 115(12): 3506–16. DOI: 10.1172/JCI24832.
66. Brickner A.G., Evans A.M., Mito J.K., et al. The PANE1 gene encodes a novel human minor histocompatibility antigen that is selectively expressed in B-lymphoid cells and B-CLL. Blood. 2006; 107(9): 3779–86. DOI: 10.1182/blood-2005-08-3501.
67. Broen K., Levenga H., Vos J., et al. A polymorphism in the splice donor site of ZNF419 results in the novel renal cell carcinoma-associated minor histocompatibility antigen ZAPHIR. PLoS ONE. 2011; 6(6): e21699. DOI: 10.1371/journal.pone.0021699.
68. Kawase T., Akatsuka Y., Torikai H., et al. Alternative splicing due to an intronic SNP in HMSD generates a novel minor histocompatibility antigen. Blood. 2007; 110(3): 1055–63. DOI: 10.1182/blood-2007-02-075911.
69. Terakura S., Murata M., Warren E.H., et al. A single minor histocompatibility antigen encoded by UGT2B17 and presented by human leukocyte antigenA*2902 and -B*4403. Transplantation. 2007; 83(9): 1242–8. DOI: 10.1097/01.tp.0000259931.72622.d1.
70. Griffi oen M., van Bergen C.A., Falkenburg J. Autosomal minor histocompatibility antigens: How genetic variants create diversity in immune targets. Front Immunol. 2016; 7: 100. DOI: 10.3389/fi mmu.2016.00100.
71. Meadows L., Wang W., den Haan J., et al. The HLA-A*0201-restricted H-Y antigen contains a posttranslationally modifi ed cysteine that signifi cantly affects T cell recognition. Immunity. 1997; 6(3): 273–81. DOI: 10.1016/s1074-7613(00)80330-1.
72. Granados D.P., Sriranganadane D., Daouda T., et al. Impact of genomic polymorphisms on the repertoire of human MHC class I-associated peptides. Nat Commun. 2014; 5: 3600. DOI: 10.1038/ncomms4600.
73. Bijen H.M., Hassan C., Kester M.G.D.G., et al. Specifi c T cell responses against minor histocompatibility antigens cannot generally be explained by absence of their allelic counterparts on the cell surface. Proteomics. 2017; 18(12): e1700250. DOI: 10.1002/pmic.201700250.
74. van Balen P., van Bergen C., van Luxemburg-Heijs S., et al. CD4 donor lymphocyte infusion can cause conversion of chimerism without GVHD by inducing immune responses targeting minor histocompatibility antigens in HLA class II. Front Immunol. 2018; 9: 3016. DOI: 10.3389/fi mmu.2018.03016.
75. Christopher M.J., Petti A.A., Rettig M.P., et al. Immune escape of relapsed AML cells after allogeneic transplantation. New Engl J Med. 2018; 379(24): 2330–41. DOI: 10.1056/nejmoa1808777.
76. Toffalori C., Zito L., Gambacorta V., et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat Med. 2019; 25(4): 603–11. DOI: 10.1038/s41591-019-0400-z.
77. Brooks A.G., Boyington J.C., Sun P.D. Natural killer cell recognition of HLA class I molecules. Rev Immunogenet. 2000; 2(3): 433–48.
78. Fuchs K.J., Honders W.M., van der Meijden E.D., et al. Optimized whole genome association scanning for discovery of HLA class I-restricted minor histocompatibility antigens. Front Immunol. 2020; 11: 659. DOI: 10.3389/fi mmu.2020.00659.
79. Martin P.J., Levine D.M., Storer B.E., et al. Genome-wide minor histocompatibility matching as related to the risk of graft-versus-host disease. Blood. 2017; 129(6): 791–798. DOI: 10.1182/blood-2016-09-737700.
80. Roy D.C., Perreault C. Major vs minor histocompatibility antigens. Blood. 2017; 129(6): 664–6. DOI: 10.1182/blood-2016-12-754515.
81. Hombrink P., Hadrup S.R., Bakker A., et al. High-throughput identifi cation of potential minor histocompatibility antigens by MHC tetramer-based screening: Feasibility and limitations. PLoS ONE. 2011; 6(8): e22523. DOI: 10.1371/journal.pone.0022523.
82. Oostvogels R., Lokhorst H., Mutis T. Minor histocompatibility Ags: Identifi cation strategies, clinical results and translational perspectives. Bone Marrow Transplant. 2015; 51(2): 163–71. DOI: 10.1038/bmt.2015.256.
83. Bleakley M., Riddell S.R. Exploiting T cells specifi c for human minor histocompatibility antigens for therapy of leukemia. Immunol Cell Biol. 2011; 89(3): 396–407. DOI: 10.1038/icb.2010.124.
84. van Bergen C.A., van Luxemburg-Heijs S.A., de Wreede L.C., et al. Selective graft-versus-leukemia depends on magnitude and diversity of the alloreactive T cell response. J Clin Invest. 2017; 127(2): 517–29. DOI: 10.1172/JCI86175.
85. Granados D., Rodenbrock A., Laverdure J-P., et al. Proteogenomic-based discovery of minor histocompatibility antigens with suitable features for immunotherapy of hematologic cancers. Leukemia. 2016; 30(6): 1344–54. DOI: 10.1038/leu.2016.22.
86. Summers C., Sheth V.S., Bleakley M. Minor histocompatibility antigen-specifi c T cells. Front Pediatr. 2020; 8: 284. DOI: 10.3389/fped.2020.00284.
87. Romaniuk D.S., Postovskaya A.M., Khmelevskaya A.A., et al. Rapid multiplex genotyping of 20 HLA-A*02:01 restricted minor histocompatibility antigens. Front Immunol. 2019; 10: 1226. DOI: 10.3389/fi mmu.2019.01226.
88. Chalmers Z.R., Connelly C.F., Fabrizio D., et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017; 9(1): 34. DOI: 10.1186/s13073-017-0424-2.
89. MacKay M., Afshinnekoo E., Rub J., et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat Biotechnol. 2020; 38(2): 233–44. DOI: 10.1038/s41587-019-0329-2.
90. Wudhikarn K., Palomba M.L., Pennisi M., et al. Infection during the fi rst year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J. 2020; 10(8): 79. DOI: 10.1038/s41408-020-00346-7
91. Mardiana S., Gill S. CAR T cells for acute myeloid leukemia: State of the art and future directions. Front Oncol. 2020; 10: 697. DOI: 10.3389/fonc.2020.00697.
92. Vago L., Perna S., Zanussi M., et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med. 2009; 361(5): 478–88. DOI: 10.1056/NEJMoa0811036.
93. Wilke M., Dolstra H., Maas F., et al. Quantifi cation of the HA-1 gene product at the RNA level; relevance for immunotherapy of hematological malignancies. Hematol J. 2003; 4(5): 315–20. DOI: 10.1038/sj.thj.6200318.
94. Nicholls S., Piper K.P., Mohammed F., et al. Secondary anchor polymorphism in the HA-1 minor histocompatibility antigen critically affects MHC stability and TCR recognition. Proc Natl Acad Sci U S A. 2009; 106(10): 3889–94. DOI: 10.1073/pnas.0900411106.
95. Fujii N., Hiraki A., Ikeda K., et al. Expression of minor histocompatibility antigen, HA-1, in solid tumor cells. Transplantation. 2002; 73(7): 1137–41. DOI: 10.1097/00007890-200204150-00022.
96. de Bueger M., Bakker A., Rood V.J., et al. Tissue distribution of human minor histocompatibility antigens. Ubiquitous versus restricted tissue distribution indicates heterogeneity among human cytotoxic T lymphocyte-defi ned non-MHC antigens. J Immunol. 1992; 149(5): 1788–94.
97. Dickinson A.M., Wang X-N., Sviland L., et al. In situ dissection of the graft-versus-host activities of cytotoxic T cells specifi c for minor histocompatibility antigens. Nat Med. 2002; 8(4): 410–14. DOI: 10.1038/nm0402-410.
98. Mutis T., Gillespie G., Schrama E., et al. Tetrameric HLA class I-minor histocompatibility antigen peptide complexes demonstrate minor histocompatibility antigen-specifi c cytotoxic T lymphocytes in patients with graft-versus-host disease. Nat Med. 1999; 5(7): 839–42. DOI: 10.1038/10563.
99. Marijt E.W., Heemskerk M.H., Kloosterboer F.M., et al. Hematopoiesisrestricted minor histocompatibility antigens HA-1- or HA-2-specifi c T cells can induce complete remissions of relapsed leukemia. Proc Natl Acad Sci U S A. 2003; 100(5): 2742–7. DOI: 10.1073/pnas.0530192100.
100. Tseng L-H., Lin M-T., Hansen J.A., et al. Correlation between disparity for the minor histocompatibility antigen HA-1 and the development of acute graftversus-host disease after allogeneic marrow transplantation. Blood. 1999; 94(8): 2911–4. DOI: 10.1182/blood.v94.8.2911.420k21_2911_2914.
101. Socié G., Loiseau P., Tamouza R., et al. Both genetic and clinical factors predict the development of graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Transplantation. 2001; 72(4): 699–706. DOI: 10.1097/00007890-200108270-00024.
102. Gallardo D., Aróstegui J., Balas A., et al. Disparity for the minor histocompatibility antigen HA 1 is associated with an increased risk of acute graft versus host disease (GvHD) but it does not affect chronic GvHD incidence, disease‐free survival or overall survival after allogeneic human leucocyte antigen identical sibling donor transplantation. Br J Haematol. 2001; 114(4): 931–6. DOI: 10.1046/j.1365-2141.2001.03013.x.
103. Murata M., Emi N., Hirabayashi N., et al. No signifi cant association between HA-1 incompatibility and incidence of acute graft-versus-host disease after HLA-identical sibling bone marrow transplantation in Japanese patients. Int J Hematol. 2000; 72: 371–5.
104. Heinemann F.M., Ferencik S., Ottinger H.D., et al. Impact of disparity of minor histocompatibility antigens HA-1, CD31, and CD49b in hematopoietic stem cell transplantation of patients with chronic myeloid leukemia with sibling and unrelated donors. Transplantation. 2004; 77(7): 1103–6. DOI: 10.1097/01.tp.0000120175.25116.cb.
105. Lin M., Gooley T., Hansen J., et al. Absence of statistically signifi cant correlation between disparity for the minor histocompatibility antigen-HA-1 and outcome after allogeneic hematopoietic cell transplantation. Blood. 2001; 98(10): 3172–3. DOI: 10.1182/blood.v98.10.3172.
106. Spellman S., Warden M.B., Haagenson M., et al. Effects of mismatching for minor histocompatibility antigens on clinical outcomes in HLA-matched, unrelated hematopoietic stem cell transplants. Biol Blood Marrow Transplant. 2009; 15(7): 856–63. DOI: 10.1016/j.bbmt.2009.03.018.
107. Teshima T., Ordemann R., Reddy P., et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002; 8(6):575–81. DOI: 10.1038/nm0602-575.
108. Matte-Martone C., Liu J., Jain D., et al. CD8+ but not CD4+ T cells require cognate interactions with target tissues to mediate GVHD across only minor H antigens, whereas both CD4+ and CD8+ T cells require direct leukemic contact to mediate GVL. Blood. 2008; 111(7): 3884–92. DOI: 10.1182/blood-2007-11-125294.
109. Miller J.S., Warren E.H., van den Brink M.R.M., et al. NCI First International Workshop on the Biology, Prevention, and Treatment of Relapse after Allogeneic Hematopoietic Stem Cell Transplantation: Report from the Committee on the Biology Underlying Recurrence of Malignant Disease following Allogeneic HSCT:Graft-versus-Tumor/Leukemia Reaction. Biol Blood Marrow Transplant. 2010; 16(5): 565–86. DOI: 10.1016/j.bbmt.2010.02.005.
110. Barge R.M.Y., Osanto S., Marijt W.A.F.E., et al. Minimal GVHD following in-vitro T cell-depleted allogeneic stem cell transplantation with reduced-intensity conditioning allowing subsequent infusions of donor lymphocytes in patients with hematological malignancies and solid tumors. Exp Hematol. 2003; 31(10):865–72. DOI: 10.1016/s0301-472x(03)00200-5.
111. Kolb H.-J. Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood. 2008; 112(12): 4371–83. DOI: 10.1182/blood-2008-03-077974.
112. Epstein F.H., Ferrara J.L.M., Deeg H.J. Graft-versus-host disease. N Engl J Med. 1991; 324(10): 667–74. DOI: 10.1056/nejm199103073241005.
113. Haan D.J., Sherman N.E., Blokland E., et al. Identifi cation of a graft versus host disease-associated human minor histocompatibility antigen. Science. 1995; 268(5216): 1476–80. DOI: 10.1126/science.7539551.
114. Pierce R.A., Field E.D., Mutis T., et al. The HA-2 minor histocompatibility antigen is derived from a diallelic gene encoding a novel human class I myosin protein. J Immunol. 2001; 167(6): 3223–30. DOI: 10.4049/jimmunol.167.6.3223.
115. Spierings E., Kim Y.-H.H., Hendriks M., et al. Multicenter analyses demonstrate signifi cant clinical effects of minor histocompatibility antigens on GvHD and GvL after HLA-matched related and unrelated hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2013; 19(8): 1244–53. DOI: 10.1016/j.bbmt.2013.06.001.
116. Heemskerk M.H., Hoogeboom M., de Paus R.A., et al. Redirection of antileukemic reactivity of peripheral T lymphocytes using gene transfer of minor histocompatibility antigen HA-2-specifi c T-cell receptor complexes expressing a conserved alpha joining region. Blood. 2003; 102(10): 3530–40. DOI: 10.1182/blood-2003-05-1524.
117. Heemskerk M.H., Hoogeboom M., Hagedoorn R., et al. Reprogramming of virus-specifi c T cells into leukemia-reactive T cells using T cell receptor gene transfer. J Exp Med. 2004; 199(7): 885–94. DOI: 10.1084/jem.20031110.
118. Bijen H.M., van der Steen D.M., Hagedoorn R.S., et al. Preclinical strategies to identify off-target toxicity of high-affi nity TCRs. Molecul Ther. 2018; 26(5):1206–14. DOI: 10.1016/j.ymthe.2018.02.017.
119. Lio H-Y., Tang J-L., Wu J., et al. Minor histocompatibility antigen HA-1 and HA-2 polymorphisms in Taiwan: Frequency and application in hematopoietic stem cell transplantation. Clin Chem Lab Med. 2010; 48(9): 1287–93. DOI: 10.1515/CCLM.2010.246.
120. Sellami M.H., Ahmed B.A., Kaabi H., et al. HA‐1 and HA‐2 minor histocompatibility antigens in Tunisians. Tissue Antigens. 2010; 75(6): 720–3.DOI: 10.1111/j.1399-0039.2010.01444.x.
121. Akatsuka Y., Nishida T., Kondo E., et al. Identifi cation of a polymorphic gene, BCL2A1, encoding two novel hematopoietic lineage-specifi c minor histocompatibility antigens. J Exp Med. 2003; 197(11): 1489–1500. DOI: 10.1084/jem.20021925.
122. Nagy B., Lundán T., Larramendy M.L., et al. Abnormal expression of apoptosis related genes in haematological malignancies: Overexpression of MYC is poor prognostic sign in mantle cell lymphoma. Br J Haematol. 2003; 120(3): 434–41. DOI: 10.1046/j.1365-2141.2003.04121.x.
123. Sochalska M., Schuler F., Weiss J.G., et al. MYC selects against reduced BCL2A1/A1 protein expression during B cell lymphomagenesis. Oncogene. 2017; 36(15): 2066–73. DOI: 10.1038/onc.2016.362.
124. Пилунов А.М., Кучмий А.А., Шитиков С.А., и др. Модификация цитотоксических лимфоцитов рецептором, специфичным к минорному антигену гистосовместимости ACC1-Y. Молекулярная биология. 2019; 53(3): 456–66. DOI: 10.1134/S0026898419030145.
125. Spierings E., Drabbels J., Hendriks M., et al. A uniform genomic minor histocompatibility antigen typing methodology and database designed to facilitate clinical applications. PLoS ONE. 2006; 1(1): e42. DOI: 10.1371/journal.pone.0000042.
126. Torikai H., Akatsuka Y., Yatabe Y., et al. Aberrant expression of BCL2A1-restricted minor histocompatibility antigens in melanoma cells: Application for allogeneic transplantation. Int J Hematol. 2008; 87(5): 467–73. DOI: 10.1007/s12185-008-0076-5.
127. Kloosterboer F.M., van Luxemburg-Heijs S.A.P., van Soest R.A., et al. Upregulated expression in nonhematopoietic tissues of the BCL2A1-derived minor histocompatibility antigens in response to infl ammatory cytokines: Relevance for allogeneic immunotherapy of leukemia. Blood. 2005; 106(12): 3955–7. DOI: 10.1182/blood-2004-09-3749.
128. Nishida T., Akatsuka Y., Morishima Y., et al. Clinical relevance of a newly identifi ed HLA A24 restricted minor histocompatibility antigen epitope derived from BCL2A1, ACC 1, in patients receiving HLA genotypically matched unrelated bone marrow transplant. Brit J Haematol. 2004; 124(5): 629–35. DOI: 10.1111/j.1365-2141.2004.04823.x.
129. Akatsuka Y., Torikai H., Inamoto Y., et al. Bone marrow may be a reservoir of long lived memory T cells specifi c for minor histocompatibility antigen. Br J Haematol. 2006; 135(3): 413–4. DOI: 10.1111/j.1365-2141.2006.06313.x.
130. Roback J.D. Vaccine-enhanced donor lymphocyte infusion (veDLI). Hematology Am Soc Hematol Educ Program. 2006; 2006(1): 486–91. DOI: 10.1182/asheducation-2006.1.486.
131. Fontaine P., Roy-Proulx G., Knafo L., et al. Adoptive transfer of minor histocompatibility antigen-specifi c T lymphocytes eradicates leukemia cells without causing graft-versus-host disease. Nat Med. 2001; 7(7): 789–94. DOI: 10.1038/89907.
132. Kloosterboer F., van Luxemburg-Heijs S., van Soest R., et al. Direct cloning of leukemia-reactive T cells from patients treated with donor lymphocyte infusion shows a relative dominance of hematopoiesis-restricted minor histocompatibility antigen HA-1 and HA-2 specifi c T cells. Leukemia. 2004; 18(4): 798–808. DOI: 10.1038/sj.leu.2403297.
133. Шитиков С.А., Кучмий А.А., Быкова Н.А. и др. In silico анализ последовательностей T-клеточных рецепторов, специфичных к минорному антигену гистосовместимости HA-2. Российский иммунологический журнал. 2019; 13(1): 31–43. DOI: 10.31857/s102872210005018-4.
134. Marijt E., Wafelman A., Hoorn M., et al. Phase I/II feasibility study evaluating the generation of leukemia-reactive cytotoxic T lymphocyte lines for treatment of patients with relapsed leukemia after allogeneic stem cell transplantation. Haematologica. 2007; 92(1): 72–80. DOI: 10.3324/haematol.10433.
135. Warren E.H., Fujii N., Akatsuka Y., et al. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specifi c for minor histocompatibility antigens. Blood. 2010; 115(19): 3869–78. DOI: 10.1182/blood-2009-10-248997.
136. Meij P., Jedema I., van der Hoorn M., et al. Generation and administration of HA-1-specifi c T-cell lines for the treatment of patients with relapsed leukemia after allogeneic stem cell transplantation: A pilot study. Haematologica. 2012; 97(8): 1205–8. DOI: 10.3324/haematol.2011.053371.
137. Bondanza A., Hambach L., Aghai Z., et al. IL-7 receptor expression identifi es suicide gene-modifi ed allospecifi c CD8+ T cells capable of self-renewal and differentiation into antileukemia effectors. Blood. 2011; 117(24): 6469–78. DOI: 10.1182/blood-2010-11-320366.
138. van der Waart A.B., van de Weem N.M.P., Maas F., et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood. 2014; 124(23): 3490–3500. DOI: 10.1182/blood-2014-05-578583.
139. Franssen L., Roeven M., Hobo W., et al. A phase I/II minor histocompatibility antigen-loaded dendritic cell vaccination trial to safely improve the effi cacy of donor lymphocyte infusions in myeloma. Bone Marrow Transplant. 2017; 52(10): 1378–83. DOI: 10.1038/bmt.2017.118.
140. Oostvogels R., Kneppers E., Minnema M., et al. Effi cacy of host-dendritic cell vaccinations with or without minor histocompatibility antigen loading, combined with donor lymphocyte infusion in multiple myeloma patients. Bone Marrow Transplant. 2016; 52(2): 228–37. DOI: 10.1038/bmt.2016.250.
141. Calmeiro J., Carrascal M.A., Tavares A.R., et al. Dendritic cell vaccines for cancer immunotherapy: The role of human conventional type 1 dendritic cells.Pharm. 2020; 12(2): 158. DOI: 10.3390/pharmaceutics12020158.
142. Vdovin A.S., Filkin S.Y., Yefi mova P.R., et al. Recombinant MHC tetramers for isolation of virus-specifi c CD8+ cells from healthy donors: Potential approach for cell therapy of posttransplant cytomegalovirus infection. Biochem Mosc. 2016; 81(11): 1371–83. DOI: 10.1134/s0006297916110146.
143. van Loenen M.M., de Boer R., van Liempt E., et al. A Good Manufacturing Practice procedure to engineer donor virus-specifi c T cells into potent antileukemic effector cells. Haematologica. 2014; 99(4): 759–68. DOI: 10.3324/haematol.2013.093690.
144. van Balen P., Jedema I., van Loenen M.M., et al. HA-1H T-Cell receptor gene transfer to redirect virus-specifi c T cells for treatment of hematological malignancies after allogeneic stem cell transplantation: A phase 1 clinical study. Front Immunol. 2020; 11: 1804. DOI: 10.3389/fi mmu.2020.01804.
145. Kretschmer L., Flossdorf M., Mir J., et al. Differential expansion of T central memory precursor and effector subsets is regulated by division speed. Nat Commun. 2020; 11(1): 113. DOI: 10.1038/s41467-019-13788-w.
146. Gamadia L.E., van Leeuwen E.M., Remmerswaal E.B., et al. The size and phenotype of virus-specifi c T cell populations is determined by repetitive antigenic stimulation and environmental cytokines. J Immunol. 2004; 172(10): 6107–14. DOI: 10.4049/jimmunol.172.10.6107.
147. Dossa R.G., Cunningham T., Sommermeyer D., et al. Development of T-cell immunotherapy for hematopoietic stem cell transplantation recipients at risk of leukemia relapse. Blood. 2018; 131(1): 108–20. DOI: 10.1182/blood-2017-07-791608.
148. Романюк Д.С., Хмелевская А.А., Постовская А.М. и др. Клинически значимые минорные антигены гистосовместимости для российских пациентов, получающих трансплантацию стволовых клеток крови. Медицинская иммунология. 2019; 21(5): 847–60. DOI: 10.15789/1563-0625-2019-5-847-860.
149. van Loenen M.M., de Boer R., Amir A.L., et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci U S A. 2010;107(24): 10972–7. DOI: 10.1073/pnas.1005802107
150. Morton L.T., Reijmers R.M., Wouters A.K., et al. Simultaneous deletion of endogenous TCRαβ for TCR gene therapy creates an improved and safe cellular therapeutic. Mol Ther. 2020; 28(1): 654–74. DOI: 10.1016/j.ymthe.2019.10.001.
151. Inaguma Y., Akahori Y., Murayama Y., et al. Construction and molecular characterization of a T-cell receptor-like antibody and CAR-T cells specifi c for minor histocompatibility antigen HA-1H. Gene Ther. 2014; 21(6): 575–84. DOI: 10.1038/gt.2014.30.
152. Sommermeyer D., Hudecek M., Kosasih P., et al. Chimeric antigen receptormodifi ed T cells derived from defi ned CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016; 30(2): 492–500. DOI: 10.1038/leu.2015.247.
153. Tantalo D.G., Oliver A.J., von Scheidt B., et al. Understanding T cell phenotype for the design of effective chimeric antigen receptor T cell therapies. J Immunother Cancer. 2021; 9(5): e002555. DOI: 10.1136/jitc-2021-002555.
Пилунов Артем Михайлович*, молекулярный иммунолог, лаборатория трансплантационной иммунологии
125167, Москва
Романюк Дмитрий Сергеевич, научный сотрудник лаборатории
трансплантационной иммунологии
125167, Москва
Ефимов Григорий Александрович, кандидат биологических наук, заведующий лабораторией трансплантационной иммунологии
125167, Москва
Савченко Валерий Григорьевич , доктор медицинских наук, профессор, академик РАН, генеральный директор
125167, Москва
Пилунов А.М., Романюк Д.С., Ефимов Г.А., Савченко В.Г. Минорные антигены гистосовместимости как мишени Т-клеточной иммунотерапии. Гематология и трансфузиология. 2021;66(3):322-345. https://doi.org/10.35754/0234-5730-2021-66-3-322-345
Pilunov A.M., Romaniuk D.S., Efimov G.A., Savchenko V.G. Minor histocompatibility antigens as targets for T-cell immunotherapy. Russian journal of hematology and transfusiology. 2021;66(3):322-345. (In Russ.) https://doi.org/10.35754/0234-5730-2021-66-3-322-345
![]()
125167, Москва, Новый Зыковский проезд, 4
ФГБУ «НМИЦ гематологии» Минздрава России
тел.: 8-926-816-3887
e-mail: o.levchenko@htjournal.ru






































