


Содержание
Перейти к:
https://doi.org/10.35754/0234-5730-2019-64-3-353-361
Перейти к:
Введение. Подкожная панникулитоподобная Т-клеточная лимфома (ППТКЛ) относится к редкой группе кожных лимфопролиферативных заболеваний с клиническими проявлениями, напоминающими панникулит, α/β-цитотоксическим иммунофенотипом опухолевых клеток и разнонаправленным течением: от индолентных до агрессивных форм.
Цель работы — описать больных ППТКЛ с агрессивным клиническим течением заболевания и рефрактерностью к нескольким линиям химиотерапии.
Результаты. Представлены два клинических наблюдения больных с генерализованным характером поражения и наличием факторов неблагоприятного прогноза, у которых достигнуты полные продолжительные ремиссии заболевания в результате применения курсов химиотерапии с включением гемцитабина.
Заключение: несмотря на то, что у обоих больных ППТКЛ наблюдалась рефрактерность как минимум к трем видам лечения, использование гемцитабина позволило достигнуть длительных полных ремиссий заболевания.
Горенкова Л.Г., Кравченко С.К., Силаев М.А., Рыжикова Н.В. ТЕРАПИЯ РЕЗИСТЕНТНЫХ ФОРМ ПОДКОЖНОЙ ПАННИКУЛИТОПОДОБНОЙ Т-КЛЕТОЧНОЙ ЛИМФОМЫ. Гематология и трансфузиология. 2019;64(3):353-361. https://doi.org/10.35754/0234-5730-2019-64-3-353-361
Gorenkova L.G., Kravchenko S.K., Silaev M.A., Ryzhikova N.V. THERAPY OF THE RESISTANT FORMS OF SUBCUTANEOUS PANNICULITIS-LIKE T-CELL LYMPHOMA. Russian journal of hematology and transfusiology. 2019;64(3):353-361. (In Russ.) https://doi.org/10.35754/0234-5730-2019-64-3-353-361
Подкожная панникулитоподобная Т-клеточная лим- фома (ППТКЛ) относится к редким типам кожных Т-клеточных лимфом, субстратом которых являются зрелые Т-лимфоциты с цитотоксическим фенотипом. Впервые новый вид Т-клеточной лимфомы, протекающий с поражением подкожной жировой клетчатки, симулирующий панникулит и с частым развитием гемофагоцитарного синдрома, был описан в 1991 г. испанскими исследователями C.L. Gonzalez и соавт. [1]. Эти так называемые «цитотоксические лимфомы» имели агрессивный характер течения, и для их лечения было необходимо проведение системной химиотерапии [2, 3]. Однако дальнейшие исследования показали, что клинические, гистологические и иммунофенотипические признаки значимо различались между лимфомами с α/β - и γ/δ-иммунофенотипами, что обусловило разделение описанного заболевания на две самостоятельные нозологические формы в классификации ВОЗ-ЕОRТС (2008 г.) [4, 5]: на подкожную панникулитоподобную Т-клеточную лимфому и первичную кожную γ/δ Т-клеточную лимфому.
Частота встречаемости заболевания составляет не более 1 % от всех неходжкинских лимфом: за 10 лет наблюдения крупными группами по изучению и лечению кожных лимфом описано не более 18 случаев [6, 8]. ППТКЛ встречается, как правило, среди больных молодого возраста (медиана возраста — 36 лет), примерно в 20 % случаев — 20 лет и моложе, чаще среди женщин (соотношение мужчины : женщины = 2,2 : 1) [7, 9]. Этиология заболевания в настоящее время не установлена, известны случаи развития ППТКЛ после органной трансплантации, иммуносупрессивной терапии [10, 11]. При изучении тропности опухолевых клеток к подкожно-жировой клетчатке выявлено наличие экспрессии на атипичных лимфоцитах CCL5 — лиганда к хемокиновым рецепторам CCR5, располагающимся на поверхности адипоцитов. Данный феномен специфичен только для ППТКЛ и отсутствует при неопухолевых поражениях подкожной жировой клетчатки, в том числе при волчаночном панникулите [12]. В 20 % случаев ППТКЛ отмечается ассоциация с аутоиммунными заболеваниями (системная красная волчанка, аутоиммунный тиреоидит, ювенильный ревматоидный артрит) [8].
Наиболее распространенными проявлениями ППТКЛ являются единичные или множественные подкожные инфильтраты на нижних конечностях, кожа над которыми может быть слегка гиперемиро- вана или не изменена [13, 14]. Болезнь протекает длительно и индолентно, в части случаев отмечается самостоятельная регрессия образований. При агрессивном течении ППТКЛ отмечается несколько иная картина, а именно — быстрый рост подкожных образований в размерах с изъязвлением, периферическая лимфаденопатия, стойкая фебрильная лихорадка, уменьшение массы тела и частое развитие жизнеугрожающего состояния — гемофагоцитарного синдрома (стойкая фебрильная лихорадка, панцитопения, гепатоспленомегалия) [15].
Диагностика ППТКЛ основана на данных гистологического, иммуногистохимического и молекулярно-генетического исследований. Патоморфологически выявляется очаговый или диффузный плотный инфильтрат, располагающийся в дольках подкожножировой клетчатки без эпидермотропизма и поражения дермы. Инфильтрат представлен плеоморфными лимфоидными клетками среднего и редко крупного размера, характерны очаги некроза подкожно-жировой клетчатки. Опухолевые клетки имеют α/β-цитотоксический иммунофенотип: экспрессируют βΡ1+, CD3+, CD8+, TIA1+, perforin+, granzyme B+, отсутствует экспрессия CD4-, CD56-, CD30-, может наблюдаться потеря пан-Т-клеточных антигенов CD2, CD5 и CD7, пролиферативная активность (Ki-67) варьирует от 10 до 90 % [16]. Молекулярно-генетические исследования определяют клональную реаранжировку генов β-цепи Т-клеточного рецептора в 100 % случаев и генов γ-цепи Т-клеточного рецептора — в 50—80 % [7, 13, 17].
Дифференциальную диагностику проводят с лимфомами, протекающими с поражением подкожножировой клетчатки, а именно с экстранодальной NK/T-клеточной лимфомой, первичной кожной анапластической крупноклеточной лимфомой и первичной кожной γ/δ -Т- клеточной лимфомой. Различие этих лимфопролиферативных заболеваний друг от друга определяется иммунофенотипом опухолевого субстрата. Однако основную сложность дифференциального диагноза составляют дерматозы с поражением подкожножировой клетчатки, самым распространенным среди которых является «люпус-панникулит». Морфологически «панникулит» имеет следующие признаки: накопление муцина, примесь плазматических клеток, скопления В-лимфоцитов с формированием центров размножения, небольшие скопления CD123 плазмацитоидных дендритических клеток [14]. Однако в части случаев подкожных панникулитоподобных лимфом, в том числе в биоптате «старого» либо частично регрессирующего очага, могут быть определены гистопатологические признаки «панникулита», аберрантный иммунофенотип и снижение индекса пролиферативной активности. Такие «перекрестные» черты ППТКЛ и «люпус-панникулита» позволили зародиться существующим и поныне двум гипотезам — трансформация доброкачественных «панникулитов» в неопластические процессы или одномоментное сосуществование Т-клеточной лимфомы и аутоиммунного заболевания [18, 19].
Прогноз при вялотекущих формах заболевания благоприятный: пятилетняя общая выживаемость составляет 80 % [8]. К факторам неблагоприятного прогноза, которые вдвое снижают медиану выживаемости больных, относят: гемофагоцитарный синдром, ангиотропизм, поражение верхних конечностей, повышение активности лактатдегидрогеназы (ЛДГ) [20].
В терапии индолентных форм ППТКЛ хорошо себя зарекомендовало использование глюкокортикостероидной или иммуносупрессивной терапии (циклоспорин, малые дозы метотрексата), которая позволяет достичь полных ремиссий заболевания; интенсивная химиотерапия в таких случаях не имеет преимуществ [9, 21, 22, 23].
При агрессивном течении заболевания и/или резистентности к вышеперечисленным лечебным опциям больные нуждаются в проведении интенсивной терапии. Согласно данным, приведенным в нескольких крупных обзорах литературы [8, 24], стандартные схемы химиотерапии с включением антрациклиновых антибиотиков недостаточно эффективны. В педиатрической практике описано успешное применение высокодозных курсов химиотерапии по протоколу NHL BFM-90 в терапии рефрактерных форм заболевания с агрессивным клиническим течением: пятилетняя общая выживаемость и бессобытийная выживаемость составили 78 и 74 %, соответственно, у 25 % больных выполнена трансплантация аутологичных гемопоэтических стволовых клеток с целью консолидации полученной ремиссии [25]. При отсутствии противоопухолевого эффекта от высокодозной химиотерапии по педиатрическим протоколам опции лечения таких больных не определены.
Цель настоящей работы — представить клинические наблюдения больных ППТКЛ с агрессивным клиническим течением заболевания и рефрактерностью к нескольким линиям химиотерапии.
В июле 2017 г. в ФГБУ «Национальный медицинский исследовательский центр гематологии» (НМИЦ гематологии) Минздрава России обратился мужчина 30 лет с жалобами на обширный язвенно-некротический дефект в надлобковой области (рис. 1а, б), множественные подкожные образования на коже груди, живота, верхних конечностей, стойкую фебрильную лихорадку 39—40 °С, уменьшение массы тела на 15— 16 кг. Впервые подкожное образование в надлобковой области появилось в марте 2017 г. и прогрессивно увеличивалось в размерах, сформировался язвенный дефект. В течение трех месяцев больной получал парентерально преднизолон в дозе 20 мг/сут. Однако эффект достигнут не был, отмечено появление новых подкожных инфильтратов. Была выполнена биопсия подкожного образования, при гистологическом исследовании которого выявлен диффузный рост лимфоидных клеток небольших размеров, на основании чего установлен диагноз Т-клеточной лимфомы. Больному был проведен курс химиотерапии по программе CHOP (схема 1), несмотря на проведение которого, отмечалась прогрессия заболевания в виде увеличения в размерах подкожных образований, появления новых.
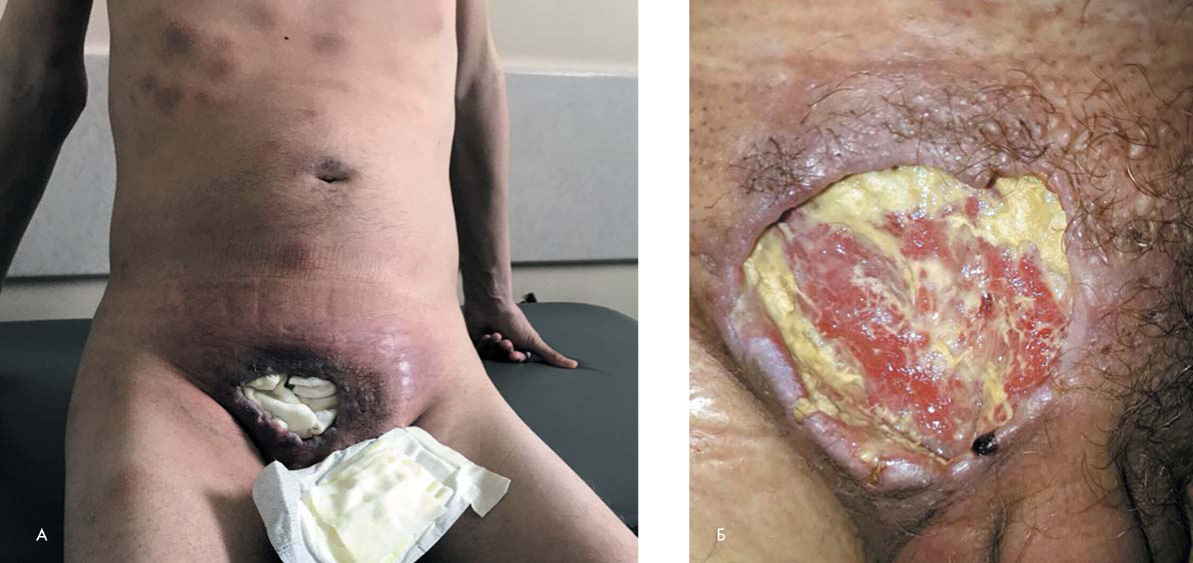
Рисунок 1. Обширный язвенно-некротический дефект в надлобковой области, А — общий вид; Б — язвенный дефект
Figure 1. A large ulcer tumor in suprapubic area, А — general view; Б — ulceralive defect
Схема 1. Программа химиотерапии CHOP
|
|
|
|
В НМИЦ гематологии произведена повторно биопсия опухоли из язвенно-некротического дефекта. При исследовании биоптата выявлена характерная гистологическая картина: диффузный плотный инфильтрат, располагавшийся в дольках подкожно-жировой клетчатки без эпидермотропизма и поражения дермы, инфильтрат был представлен плеоморфными лимфоидными клетками среднего размера, присутствовали очаги некроза подкожно-жировой клетчатки. На основании гистологической картины, α/β-цитотоксического иммунофенотипа и выявленной клональной реаранжировки генов β-цепи Т-клеточного рецептора верифицирована ППТКЛ.
Несмотря на отсутствие гемофагоцитарного синдрома, болезнь протекала агрессивно, отмечалось генерализованное поражение в виде инфильтратов в подкожно-жировой клетчатке на туловище и верхних конечностях, В-симптомы, повышение сывороточной активности ЛДГ. Течение ППТКЛ было резистентно к терапии глюкокортикостероидными гормонами (первая линия) и химиотерапии по схеме CHOP (схема 1) (вторая линия).
При выборе метода лечения в ФГБУ «НМИЦ гематологии» было принято решение о проведении блоковой терапии по протоколу NHL BFM-90: выполнен блок А (схема 2), в посткурсовом интервале возникли серьезные осложнения: длительный глубокий миелотоксический агранулоцитоз, грамотрицательный сепсис, вызванный Escherichia coli, грам- положительная катетер-ассоциированная инфекция кровотока (Staphylococcus aureus), мукозит 3-й степени, некротическая энтеропатия, печеночная недостаточность. В результате лечения было достигнуто незначительное улучшение в виде небольшого уменьшения в размерах язвенно-некротического дефекта. После окончания второго высокодозного курса химиотерапии по выбранной программе (блок С) отмечено вновь появление В-симптомов, новых подкожных образований, что свидетельствовало о рефрактерности опухоли к проводимой терапии.
Схема 2. Протокол NHL BFM-90
Блок А: дексаметазон 10 мг/м2 в/в капельно 1-5-й день; винкристин 1,4 мг/м2 в/в струйно 1-й день (не более 2 мг); метотрексат 1000 мг/м2 в/в капельно за 24 часа, кальция фолинат вводится через 12 часов от окончания введения метотрексата; цитатрабин 150 мг/м2 в/в капельно каждые 12 часов 4-5-й день; этопозид 100 мг/м2 в/в капельно 4-5-й день; ифосфамид 800 мг/м2 в/в капельно 1-5-й день (обязательно месна — во время инфузии, через 4 и 8 часов после окончания введения ифосфамида, из расчета 60 % от дозы вводимого ифосфамида). Блок С: дексаметазон 20 мг/м2 в/в капельно 1 -5-й день; винорельбин 20 мг в/в струйно; цитозар 2 г/м2 каждые 12 часов 3-часовая инфузия в/в капельно 1-2-й день; вепезид 150 мг/м2 в/в капельно 3-5-й день. |
Следующей линией лечения была выбрана химиотерапия с включением гемцитабина, учитывая потенциальную эффективность этого цитостатического препарата при лечении рефрактерных нодальных Т-клеточных лимфом. После одного курса по программе ESGAP (схема 3) отмечена полная регрессия фебрильной лихорадки, всех инфильтратов в подкожно-жировой клетчатке в области груди, живота и верхних конечностей и постепенное сокращение в размерах язвы надлобковой области. Переносимость лечения была удовлетворительной. Суммарно выполнено три цикла химиотерапии по выбранной программе. По данным позитронно-эмиссионного исследования, совмещенного с компьютерной томографией (ПЭТ/КТ), после окончания третьего курса химиотерапии сохранялось лишь небольшое накопление радиофармпрепарата в инфильтрате подкожно-жировой клетчатки надлобковой области, что соответствовало 3 баллам по шкале Deauville (DS) [26]. С целью консолидации частичной ремиссии заболевания выполнена трансплантация аутологичных гемопоэтических стволовых клеток крови. При контрольном ПЭТ/КТ исследовании отмечено достижение полного метаболического ответа (DS 1 балл).
Схема 3. Программа химиотерапии ESGAP
солумедрол 500 мг/ м2 в/в кап 1-5-й день; этопозид 100 мг в/в кап 1-4-й день; цисплатин 25 мг/ м2 в/в кап за 8 часов 1-4-й день; гемцитабин 1000 мг/м2 в/в кап за 1 час 5-й день. |
На настоящий момент срок наблюдения составляет 12 месяцев: сохраняется полная ремиссия заболевания: заживление и рубцевание язвенного дефекта в надлобковой области, отсутствие образований в подкожно-жировой клетчатке ранее выявляемых и других локализациях (рис. 2, 3).
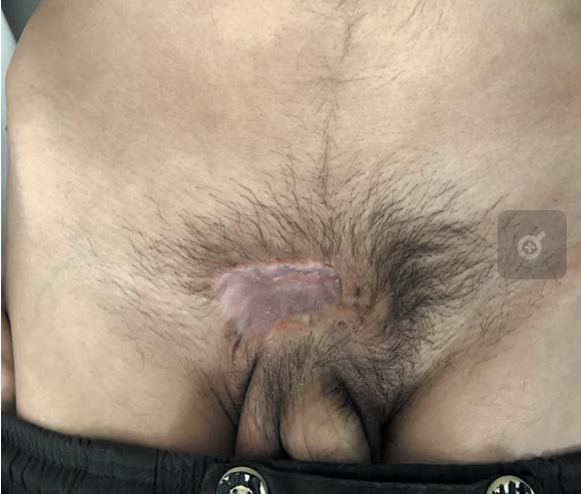
Рисунок 2. Заживление и рубцевание язвенного дефекта в надлобковой области
Figure 2. Regression and scarring of the ulcer in the suprapubic area
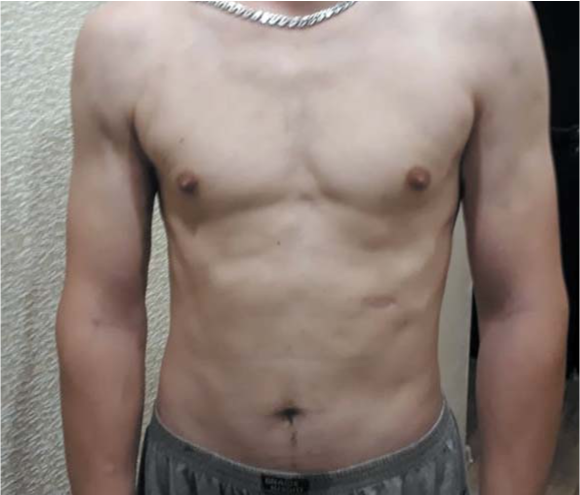
Рисунок 3. Отсутствие образований подкожно-жировой клетчатки в ранее выявляемых и других локализациях
Figure 3. The absence of subcutaneous tumor in previously detected and other localizations
В НМИЦ гематологии обратилась женщина 36 лет с жалобами на множественные подкожные образования передней стенки брюшной полости, поясничной и ягодичной областей. Диагноз ППТКЛ был верифицирован у нее весной 2018 г. на основании анамнеза и клинической картины заболевания, инструментальных методов обследования, гистологического исследования биоптата инфильтрата подкожно-жировой клетчатки, при котором выявлен инфильтрат, состоящий из лимфоидных клеток небольших размеров, располагающийся вдоль адипоцитов, а также иммуногистохимического исследования биоптата — иммунофенотип опухолевых клеток был представлен экспрессией CD3, CD8, TIA1, perforin, granzyme B, отсутствовала экспрессия CD4, CD56. Кроме того, была выявлена клональная реарран- жировка генов и γ-цепи Т-клеточного рецептора.
Больной была проведена первая линия терапия ППТКЛ — циклоспорин А в течение месяца, однако противоопухолевого эффекта достигнуто не было. Следующим этапом лечения было проведение курса системной химиотерапии по программе CHOP, в результате которого также не отмечено положительной динамики.
Учитывая агрессивный характер заболевания, прогрессию после терапии циклоспорином А в течение одного месяца, химиотерапии по программе CHOP, больной было выполнено четыре курса высокодозной химиотерапии (суммарно в течение 2,5 месяца): цисплатин 25 мг/м2, цитарабин 2 г/м2, этопозид 100 мг/м2, метилпреднизолон 500 мг/м2. В результате был достигнут противоопухолевый эффект в виде регрессии всех выявляемых элементов. Однако через две недели после окончания четвертого курса химиотерапии отмечено появление двух новых подкожных образований на боковой поверхности брюшной стенки справа, в которых, по данным ПЭТ/КТ, отмечалось специфическое накопление радиофармпрепарата (SUVmax 7,8). Для верификации нозологической формы заболевания была выполнена биопсия одного из образований и на основании гистологического исследования подтвержден диагноз ППТКЛ.
В качестве четвертой линии лечения проведена терапия с включением гемцитабина (схема 4). По данным контрольного ПЭТ/КТ, после 4-го цикла терапии достигнута ремиссия заболевания.
Схема 4. Программа химиотерапии IGEV
дексаметазон 40 мг в/в 1-5-й день; |
ифосфамид 2 г/ м2 в/в кап 1-4-й день; |
винорельбин 20 мг/ м2 в/в 1 -й день; |
гемцитабин 800 мг/м2 в/в кап 1 -й, 5-й день. |
ППТКЛ является крайне редкой нозологической формой среди кожных лимфом, развивается преимущественно в молодом возрасте и менее чем в 20 % случаев ассоциируется с агрессивным характером течения заболевания [13, 14]. В представленных клинических наблюдениях больные были моложе 40 лет, к моменту появления первых признаков заболевания, отмечалось быстротекущее течение болезни с генерализованным характером поражения.
У больных имелись множественные подкожные образования на животе, груди, верхних и нижних конечностях, кожа над инфильтратами могла быть не изменена, эритематозна или иметь вид экхимоза, у первого больного в надлобковой области определялся обширный (более 12 см в диаметре) язвенно-некротический дефект.
В типичных случаях диагностика лимфомы характеризуется морфогистологическими признаками, такими как наличие очагового или диффузного плотного инфильтрата, располагающегося в дольках подкожно-жировой клетчатки, без эпидермотропизма и поражения дермы; плеоморфными лимфоидными клетками среднего и редко крупного размера с иммунофенотипом βΡ1+, CD3+, CD8+, CD4-, TIA1+, perforin+, granzyme B+, CD56-, CD30-. Выявление Т-клеточной клональности по реарранжировке генов Т-клеточного рецептора является важным дополнительным методом диагностики ППТКЛ. У 2 больных выполнены повторные биопсии кожи и подкожно-жировой клетчатки и выявлены характерные гистологические признаки и иммунофенотип опухолевых клеток, а также определена моноклональность по генам β-цепи (в 1-м случае) и γ-цепи ТКР (во 2-м случае).
Несмотря на благоприятный прогноз при данном заболевании, информацию о хороших результатах лечения глюкокортикостероидными гормонами и иммуносупрессивной терапией по литературным данным [8], в представленных клинических наблюдениях отмечалось рефрактерное течение к описанной терапии, а также к химиотерапии по программе СНОР.
К факторам неблагоприятного прогноза относят гемофагоцитарный синдром, поражение подкожно-жировой клетчатки верхних конечностей, повышение активности ЛДГ. Хотя развитие гемофагоцитарного синдрома не было отмечено ни в одном случае, два других прогностически неблагоприятных признака присутствовали у обоих больных.
Терапия по программе CHOP для агрессивных форм ППТКЛ не приводит к достижению высоких результатов, но в то же время отсутствуют стандарты высокодозной химиотерапии. У первого больного резистентность отмечалась и к высокодозной химиотерапии по протоколу NHL BFM-90, во 2-м случае — к курсам, которые используются для терапии 2-й линии диффузных В-клеточных крупноклеточных лимфом.
Схемы лечения с применением гемцитабина хорошо зарекомендовали себя в терапии периферических Т-клеточных лимфом в первой и последующих линиях [27, 28]. Как показывают результаты лечения больных в представленных наблюдениях, несмотря на рефрактерность, как минимум к трем видам лечения, использование гемцитабина позволило достигнуть длительных полных ремиссий заболевания.
1. Gonzalez C.L., Medeiros L.J., Braziiel R.M., Jaffe E.S. T-cell lymphoma involving subcutaneous tissue: a clinicopathologic entity commonly associated with hemophagocytic syndrome. Am J Surg Pathol. 1991; 15(1): 17–27.
2. Hoque S.R., Child F.J., Whittaker S.J. et al. Subcutаneous panniculitis-like T-cell lymphoma: a clinicopathological, immunophenotypic and molecular analysis of six patients. Br J Dermatol. 2003; 148 (3): 516–25.
3. Lozzi G.P., Massone C., Citarella L. et al. Rimming of adipocytes by neoplastic lymphocytes:a histopathologic feature not restricted to subcutaneous T-cell lymphoma. Am J Dermatopathol. 2006; 28: 9–12.
4. Willemze R., Cerroni L., Kempf W. et al. The 2018 update of the WHO-EORTC classifi cation for primary cutaneous lymphomas. Blood. 2019; 133 (16): 1703– 14. DOI: 10.1182/blood-2018-11-881268
5. Swerdlow S.H., Campo E., Pileri S.A. et al. The 2016 revision of the World Health Organization classifi cation of lymphoid neoplasms. Blood. 2016; 127: 2375–90.
6. Lopez-Lerma I., Penate Y., Gallardo F. Subcutaneous panniculitis-like T-cell lymphoma: clinical features, therapeutic approach, and outcome in a case series of 16 patients. J Am Acad Dermatol. 2018; 79(5): 892–8.
7. Rutnin S., Porntharukcharoen S., Boonsakan P. Clinicopathologic, immunophenotypic, and molecular analysis of subcutaneous panniculitis-like T-cell lymphoma: A retrospective study in a tertiary care center. J Cutan Pathol. 2019; 46 (1): 44–51.
8. Willemze R., Jansen P.M., Cerroni L. et al. Subcutaneous panniculitis-like T-cell lymphoma: defi nition, classifi cation, and prognostic factors: an EORTC Cutaneous Lymphoma Group study of 83 cases. Blood. 2008; 111(2): 838–45.
9. Michonneau D., Petrella T., Ortonne N. et al. Subcutaneous panniculitis-like T-cell lymphoma: immunosuppressive drugs induce better response than polychemotherapy. Acta Derm Venereol. 2017; 97(3): 358–64.
10. Michot C., Costes V., Gerard-Dran D. et al. Subcutaneous panniculitis-like T-cell lymphoma in a patient receiving etanercept for rheumatoid arthritis. Br J Dermatol. 2009; 160: 889–90.
11. Bregman S.G., Yeaney G.A.,Greig B.W. et al. Subcutaneous panniculitislike T-cell lymphoma in a cardiac allograft recipient. J Cutan Pathol. 2005; 32: 366–70.
12. Margo C.M., Wang X. CCL5 expression in panniculitic T-cell dyscrasias and its potential role in adipocyte tropism. Am J Dermatopathol. 2013; 35: 332–7.
13. Kong Y.Y., Dai B., Kong J.C. et al. Subcutaneous panniculitis-like T-cell lymphoma: a clinicopathologic, immunophenotypic and molecular study of 22 Asian cases according to WHO-EORTC classifi cation. Am J Surg Pathol. 2008; 32 (10): 1495–502.
14. Ohtsuku M., Miura T., Yamamoto T. Clinical characteristics, differential diagnosis, and treatment outcome of subcutaneous panniculitis-like T-cell lymphoma: a literature review of published Japanese cases. Eur J Dermatol. 2017; 27(1): 34–41.
15. Yi L., Qun S., Wenjie Z. et al. The presenting manifestations of subcutaneous panniculitis-like T-cell lymphoma and T-cell lymphoma and cutaneous γ/δ T-cell ymphoma may mimic those of rheumatic diseases: a report of 11 cases. Clin Rheumatol. 2013; 32: 1169–75.
16. Willemze R. Cutaneous lymphomas with a panniculitic presentation. Seminars in Diagnostic Pathology. 2017; 34(1): 36–43. DOI: 10.1053/j.semdp.2016.11.009.
17. Lee D.W., Yang J.H., Lee S.M. et al. Subcutaneous panniculitis-like T-cell lymphoma: a clinical and pathologic study of 14 Korean patients. Ann Dermatol. 2011; 23(3): 329–37.
18. Sitthinamsuwan P, Pattanaprichakul P., Treetipsatit J. et al. Subcutaneous Panniculitis-Like T-Cell Lymphoma Versus Lupus Erythematosus Panniculitis: Distinction by Means of the Periadipocytic Cell Proliferation Index. Am J Dermatopathol. 2018; 40(8): 567–74.
19. Pincus L.B., LeBoit P.E., McCalmont T.H. et al. Subcutaneous panniculitis-like T-cell lymphoma with overlapping clinicopathologic features of lupus erythematosus: coexistence of 2 entities. Am J Dermatopayhol. 2009; 31: 520–6.
20. Massone C., Lozzi G.P., Egberts F. et al. The protean spectrum of non-Hodgkin lymphomas with prominent involvement of subcutaneous fat. J. Cutan. Pathol. 2006; 33: 418–25.
21. Mizutani S., Kuroda J., Shimura Y. et al. Cyclosporine A for chemotherapyresistant subcutaneous panniculitis-like T cell lymphoma with hemophagocytic syndrome. Acta Haematol. 2011; 126: 8–12.
22. Jang M.S., Baek J.W., Kang D.Y. et al. Subcutaneous panniculitis-like T-cell lymphoma: successful treatment with systemic steroid alone. J. Dermatol. 2012; 39(1): 96–9.
23. Briki H., Bouaziz J.D., Molinier-Frenkel V. et al. Subcutaneous panniculitis-like T-cell lymphoma: complete sustained remission with corticosteroids and methotrexate. Br J Dermatol. 2010; 163(5): 1136–8.
24. Go R.S., Wester S.M. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systemic analysis of 156 patients reported in literature. Cancer. 2004; 101(6): 1404–13.
25. Mellgren K., Attarbaschi A., Abla O. et al. Non-anaplastic peripheral T-cell lymphoma in children and adolescents — an International review of 143 cases. Ann Hematol. 2016; 95(8): 1295–305.
26. Cheson B.D. PET/CT in lymphoma: Current overview and future directions. Semin Nucl Med, 2018; 48(1): 76–81.
27. Ng M., Waters J., Cunningham D. et al. Gemcitabine, cisplatin and methylprednisolone (GEM-P) is an effective salvage regimen in patients with relapsed and refractory lymphoma. Br J Cancer. 2005; 92(8): 1352–7.
28. Yim K.L., Ashley S. Assessment of gemcitabine, cisplatin and methylprednisolone (GEM-P) combination treatment for non-Hodgkin T cell lymphoma. Med Oncol. 2012; 29(5): 3535–9.
кандидат медицинских наук, научный сотрудник отделения интенсивной высокодозной химиотерапии гемобластозов с круглосуточным стационаром,
125167, г. Москва, Новый Зыковский проезд, 4
кандидат медицинских наук, доцент, заведующий отделением интенсивной высокодозной химиотерапии гемобластозов с круглосуточным стационаром,
125167, г. Москва, Новый Зыковский проезд, 4
кандидат медицинских наук, врач-хирург научно-клинического отделения гематологической хирургии,
125167, г. Москва, Новый Зыковский проезд, 4
научный сотрудник лаборатории молекулярной гематологии,
125167, г. Москва, Новый Зыковский проезд, 4
Горенкова Л.Г., Кравченко С.К., Силаев М.А., Рыжикова Н.В. ТЕРАПИЯ РЕЗИСТЕНТНЫХ ФОРМ ПОДКОЖНОЙ ПАННИКУЛИТОПОДОБНОЙ Т-КЛЕТОЧНОЙ ЛИМФОМЫ. Гематология и трансфузиология. 2019;64(3):353-361. https://doi.org/10.35754/0234-5730-2019-64-3-353-361
Gorenkova L.G., Kravchenko S.K., Silaev M.A., Ryzhikova N.V. THERAPY OF THE RESISTANT FORMS OF SUBCUTANEOUS PANNICULITIS-LIKE T-CELL LYMPHOMA. Russian journal of hematology and transfusiology. 2019;64(3):353-361. (In Russ.) https://doi.org/10.35754/0234-5730-2019-64-3-353-361
![]()
125167, Москва, Новый Зыковский проезд, 4
ФГБУ «НМИЦ гематологии» Минздрава России
тел.: 8-926-816-3887
e-mail: o.levchenko@htjournal.ru






































