

ОРИГИНАЛЬНЫЕ СТАТЬИ

Введение. Болезнь Виллебранда (БВ), являющаяся одной из самых распространенных коагулопатий, имеет сложный характер наследования, который, в зависимости от типа заболевания, может быть как доминантным, так и рецессивным.
Цель настоящей работы — сопоставление клинических, коагулогических и молекулярно-генетических данных, полученных при обследовании больных различными типами БВ. Материалы и методы: экзоны гена vWF для 16 больных БВ секвенировали по методу Сэнгера.
Результаты. Всего было выявлено 12 различных мутаций, одна из которых (Pro2527His) ранее в мировой популяции не встречалась. Наиболее распространенной оказалась микроделеция c.2435delC, являющаяся мажорной во многих странах Европы. Она встретилась у 9 больных, 6 из которых имели самый тяжелый рецессивный 3-й тип заболевания (3 гомозиготы). Еще у 2 больных это нарушение сочеталось с миссенс-мутацией Thr791Met, что позволило диагностировать у них достаточно редкий рецессивный вариант БВ — 2N. В целом, данные молекулярно-генетического анализа соответствовали результатам дифференциальной диагностики типа БВ, основанной на клинической картине заболевания и коагулогических характеристиках. Только в одном случае у больной с предполагаемым 1-м типом БВ была выявлена мутация Arg1374Cys, характерная для типа 2 (A/M). Большая часть мутаций была обнаружена в экзонах 18 (преимущественно это была делеция c.2435delC) и 28, что делает эти экзоны наиболее перспективными при поиске мутаций.
Заключение. Начинать поиск мутаций в гене vWF целесообразно с экзонов 18 и 28. Полученные данные могут послужить основой для создания экономичного алгоритма поиска мутаций в гене vWF у отечественных больных БВ.
Введение. Прогноз лимфомы из клеток мантии (ЛКМ) определяется не только интенсификацией первой линии терапии, но и биологическими характеристиками опухоли.
Цель: оценить частоту встречаемости и выживаемость больных ЛКМ с мутациями в гене TP53.
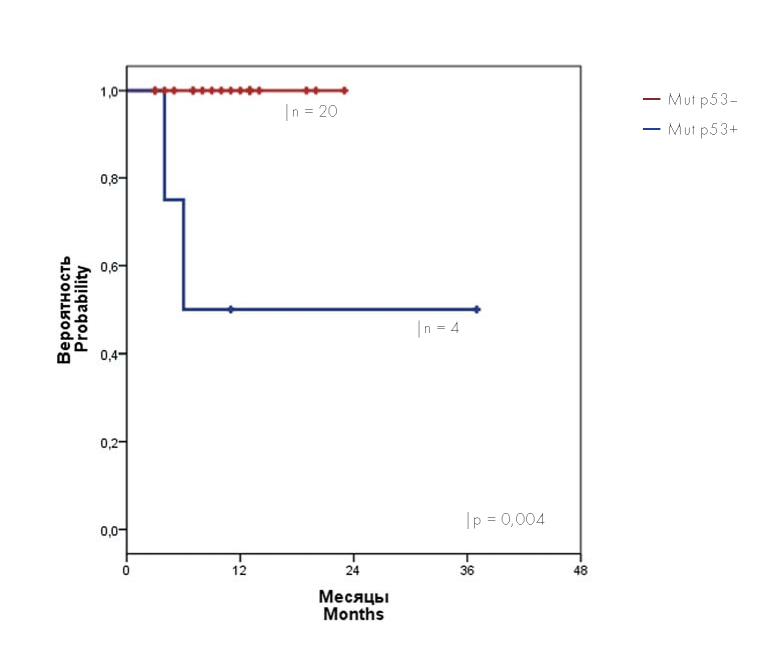
Материалы и методы. В рамках проспективного исследования «ЛКМ-2016» с января 2016 г. по декабрь 2018 г. проведено исследование по определению мутации гена TP53 у 24 больных с ЛКМ. Скрининг мутаций гена ТР53 проводили с помощью секвенирования ДНК по Сэнгеру (экзоны 1(2)–11). Идентифицировали 20 больных без мутаций в гене TP53 (20 mut p53–) и 4 — с мутацией TP53 (4 mut p53+).
Результаты. 17 больным ЛКМ (mut p53–) было проведено два цикла R-BAC (ритуксимаб, бендамустин, цитарабин) и два цикла R-HA (ритуксимаб, цитарабин 12 г/м2 ) с последующей трансплантацией аутологичных гемопоэтических стволовых клеток (ауто-ТГСК). После окончания терапии у всех 17 больных минимальная остаточная болезнь (МОБ) не определялась, у 3 больных продолжается терапия. Все больные, которые полностью завершили терапию, остаются в полной ремиссии с медианой наблюдения пять месяцев после ауто-ТГСК (от 1 до 17 месяцев). У больных ЛКМ с мутациями TP53 прогноз был хуже. Несмотря на использование интенсивной терапии, 2 больных с мутациями TP53 в данном исследовании умерли от прогрессии ЛКМ. Двум больным с мутацией TP53 была выполнена трансплантация аллогенных гемопоэтических стволовых клеток (алло-ТГСК) от неродственных полностью идентичных доноров. При сроках наблюдения 3 и 27 месяцев после алло-ТГСК больные живы, сохраняется полная ремиссия.
Заключение. Программа R-BAC/R-HA позволила достичь полной клинической и МОБ-негативной ремиссии у всех больных в группе mut p53–, с приемлемым профилем токсичности. Для больных ЛКМ с мутациями гена TP53 аллоТГСК является единственным вариантом излечения.
Введение. Интенсивное развитие радиокоммуникаций и электрокоммуникаций, различных электронных устройств приводит к «электромагнитному загрязнению» окружающей среды.
Целью исследования явилось изучение показателей железа в сыворотке крыс, подвергшихся воздействию хронического электромагнитного излучения (ЭМИ) дециметрового диапазона.
Материалы и методы. Исследование проведено на крысах, которые были разделены на экспериментальную и контрольную группы. Экспериментальная группа подразделялась на 4 подгруппы по 10 животных в каждой, которые подвергались воздействию ЭМИ (частота 460 МГц, аппарат «Волна-2») в течение 1, 2, 3 и 4 недель. Контрольную группу (10 крыс) подвергали «ложному» облучению при выключенном аппарате. Оценивали сывороточное железо (СЖ), общую железосвязывающую способность (ОЖСС) и ненасыщенную железосвязывающую способность (НЖСС) сыворотки, насыщение трансферрина железом (НТЖ), сывороточные концентрации трансферрина, гаптоглобина, малонового диальдегида, гидроперекисей липидов.
Результаты. Различия концентраций СЖ с контрольной группой (30,5 ± 3,3 мкмоль/л) обнаружены в подгруппах животных, облученных в течение 3 и 4 недель (44,1 ± 3,1 и 56,8 ± 4,4 мкмоль/л соответственно). ОЖСС у опытных животных увеличилась на 41 % (p < 0,05) по сравнению с контрольной группой (110,8 ± 10,1 мкмоль/л) только после 3 недель облучения (156,2 ± 18,2 мкмоль/л), на 4-й неделе отмечено уменьшение ОЖСС до 123,6 ± 16,4 мкмоль/л. Концентрация трансферрина повысилась с 45,6 ± 8,0 мкмоль/л в контроле до 81,0 ± 11,5 мкмоль/л на 3-й неделе облучения, на 4-й неделе отмечено ее уменьшение до 55,9 ± 6,7 мкмоль/л. НТЖ увеличилось с 27,5 % в контроле до 45,9 % только после 4 недель облучения. Содержание гидроперекисей липидов и малонового диальдегида в крови у облученных крыс было выше по сравнению с контрольными животными. Концентрация гаптоглобина в сыворотке была 26,7 % в контрольной группе, 53,8 мг % после 3 недель и 47,8 мг % после 4 недель облучения.
Заключение. ЭМИ дециметрового диапазона при тотальном хроническом облучении оказывает окислительное действие на организм.

Введение. Поражение почек в дебюте множественной миеломы (ММ) имеет место у 20–40 % больных, что в 2–4 % случаев требует начала проведения заместительной почечной терапии. Ухудшение функции почек ассоциируется с высоким риском ранней смерти, частыми осложнениями и ухудшением качества жизни.
Цель представленной работы — анализ лечения больных впервые диагностированной ММ, осложненной тяжелой и диализ-зависимой почечной недостаточностью.
Материалы и методы. В ретроспективное исследование (11.2014–11.2017) включено 62 больных ММ со скоростью клубочковой фильтрации <30 мл/мин/1,73 м2. Критерии включения — концентрация свободных легких цепей в сыворотке крови >500 мг/л и селективный характер протеинурии. Критерий исключения — диагностированный AL-амилоидоз. В зависимости от необходимости проведения гемодиализа больные были разделены на две группы: (I) не нуждающиеся в нём (n = 16) и (II) диализ-зависимые больные (n = 46).
Результаты. В индукции использовали бортезомиб-содержащие программы: VCD — 41 (66,1 %), PAD — 2 (3,2 %), VD — 12 (19,4 %) и VMP — 7 (11,3 %). Высокодозная консолидация с трансплантацией аутологичных гемопоэтических стволовых клеток реализована у 10 (16,1 %) больных. Частота общего противомиеломного ответа по группам составила 64,3 и 85,3 % (р = 0,047), включая полные и строгие полные ремиссии в 14,3 и 14,7 % случаев. Почечного ответа достигли 57,2 и 23,5 % (р = 0,032) больных. При медиане наблюдения 32,1 мес. по всей когорте медиана выживаемости без прогрессирования (ВБП) составила 14,5 мес., 3-летняя ВБП — 27,4 ± 6,6 %, медиана общей выживаемости (ОВ) — 33,6 мес. и 3-летняя ОВ — 41,5 ± 7,7 %. Различий между группами сравнения по показателям выживаемости нет. По выборке оцененных больных (n = 48) достижение любого почечного ответа ассоциировалось с улучшением показателей 3-летней ВБП 61,1 ± 11,5 % против 17,7 ± 7,7 % (р = 0,045), ОВ 72,2 ± 10,6 % против 38,1 ± 10,4 % (р = 0,069). Предиктором почечного ответа оказалось время, прошедшее между первой процедурой гемодиализа и стартом противомиеломной химиотерапии. В группе больных, достигших почечного ответа, среднее время составило 8,6 (95 % доверительный интервал 3,5–13,7) дня против 42,5 (12,6–72,5) дня больных без ответа (p = 0,045).
Заключение. Применение схем на основе бортезомиба обеспечивает высокую частоту противоопухолевых ответов с вероятностью прекратить диализ у 23,5 % зависимых больных. К возможным причинам низкой частоты почечного ответа можно отнести позднюю диагностику ММ как причины поражения почек и отсутствие доступа к новым противомиеломным препаратам в случае необходимости смены индукционной терапии.
Введение. Ротационная тромбоэластометрия (РОТЭМ) является методом исследования гемостаза, который выполняется по месту лечения и позволяет выявлять нарушения по внешнему и/или внутреннему пути свертывания крови.
Цель работы — изучить возможность использования РОТЭМ для диагностики дефицита отдельных факторов свертывания крови и мониторинга эффективности и безопасности гемостатической терапии, проводимой при этих коагулопатиях.
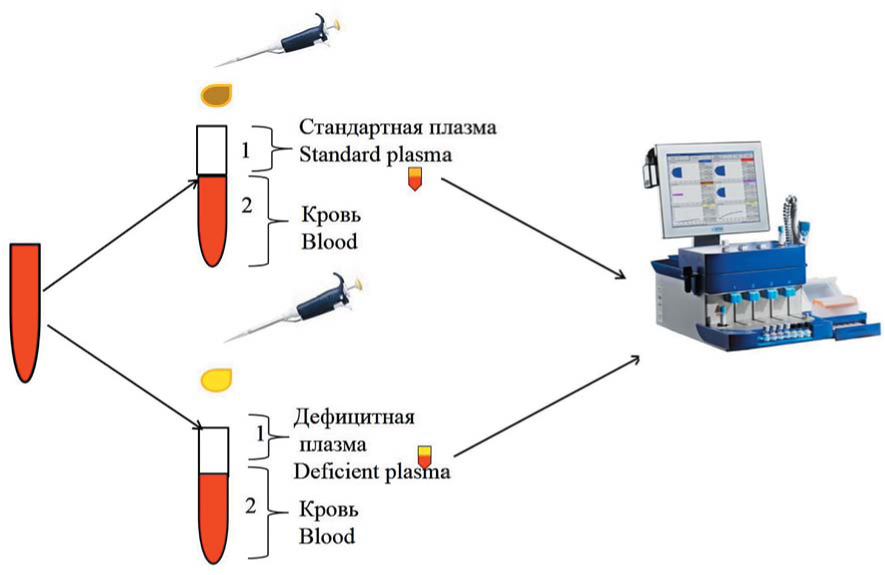
Материалы и методы. В исследование было включено 9 больных с врожденным дефицитом различных факторов свертывания крови. Проводили коагулогические тесты (определение активированного частичного тромбопластинового времени, протромбина по Квику, фибриногена, плазменной активности факторов свертывания FV, FVII, FVIII, FIX, FXI и FXII) и РОТЭМ. Для исключения влияния гепарина или гипофибриногенемии выполняли HEPTEM и/или FIBTEM. Для выявления дефицита отдельных факторов свертывания крови использовали образцы стандартной плазмы и плазмы, дефицитной по одному из факторов свертывания крови. Исследуемую цитратную кровь смешивали в соотношении 2:1 со стандартной плазмой и с дефицитной по одному из исследуемых факторов плазмой, повторно выполняли тесты EXTEM и/или INTEM. Дефицит фактора свертывания крови подтверждали по изменению параметра CT.
Результаты. Изолированное удлинение CT в тесте EXTEM выявлялось при дефиците FVII, удлинение CT только в INTEM возникало при дефиците FVIII, FIX, FXI, FXII, одновременное удлинение EXTEM и INTEM определялось при дефиците FV. При выполнении пробы со смесью цитратной крови и стандартной плазмы параметр CT укорачивался в тесте EXTEM при дефиците FVII, в тесте INTEM — при дефиците FVIII, FIX, FXI, FXII, в обоих тестах — при дефиците FV. В пробе со смесью цельной крови и дефицитной по исследуемому фактору плазмой интервал CT сохранялся удлиненным либо увеличивался. Приведены клинические примеры диагностики дефицита факторов свертывания с помощью РОТЭМ.
Заключение. Нормализация показателей РОТЭМ в пробе со стандартной плазмой и сохранение гипокоагуляции в пробе с дефицитной по фактору плазмой позволяет подтвердить дефицит фактора свертывания крови. С помощью РОТЭМ возможно проведение мониторинга эффективности гемостатической терапии при врожденных дефицитах отдельных факторов свертывания крови.

Введение. CD4+ Т-лимфоцитопения относится к общеизвестным проявлениям нарушений иммунитета при классической лимфоме Ходжкина (кЛХ), однако в существующих прогностических моделях в качестве фактора риска не учитывается. В исследованиях у онкологических больных была установлена ассоциация СD4+ Т-лимфоцитопении (<0,2 × 109 /л) с выраженным сокращением ожидаемой продолжительности жизни.
Цель: оценить влияние исходного дефицита CD4+ Т-лимфоцитов в периферической крови на общую выживаемость (ОВ) и выживаемость без прогрессирования (ВБП) у больных кЛХ.
Материалы и методы. Проведен ретроспективный анализ прогностической значимости сниженного содержания CD4+ Т-лимфоцитов в периферической крови до начала лечения у 162 больных кЛХ, которые получали комбинированную терапию в МРНЦ им. А.Ф. Цыба в период с 2000 по 2016 г. Выделяли умеренный (0,2–0,4 × 109 /л) и глубокий (<0,2 × 109 /л) дефицит CD4+ Т-лимфоцитов.
Результаты. Умеренная и глубокая CD4+ -лимфопения наблюдались соответственно у 36 (22 %) и 24 (15 %) из 162 больных. Содержание CD4+ -лимфоцитов <0,2 × 109 /л ассоциировалось с возрастом ≥45 лет (p = 0,063), продвинутой стадией (p = 0,03) и международным прогностическим индексом (МПИ) ≥4 (p = 0,000). При медиане наблюдения 72 месяца у больных с исходным количеством CD4+ Т-лимфоцитов ≤0,4 × 109 /л отмечено снижение ВБП и ОВ по сравнению с больными без CD4+ -лимфопении. В группе с неблагоприятной I–II стадией (n = 29) у 6 больных с CD4+ -лимфопенией ВБП составила 50 % против 95 % (p = 0,001), а ОВ — 40 % против 100 % (p = 0,000). В группе c кЛХ III–IV стадий (n = 120) у 53 больных с CD4+ -дефицитом ВБП и ОВ составили соответственно 57 % против 83 % (p = 0,002) и 75 % против 98 % (р = 0,004). В благоприятной прогностической подгруппе больных III–IV стадий с МПИ 0–3 (n = 94) у 32 (34 %) больных с CD4+ -дефицитом отмечены пятилетняя ВБП 66 % против 84 % (p = 0,037) и ОВ 84 % против 100 % (p = 0,117).
Заключение. Ассоциация CD4+ -дефицита с неудачами лечения больных кЛХ важна при ранней стадии заболевания, а также в подгруппе больных III–IV стадий с МПИ 0–3. Представляются оправданными модификации рискадаптированной терапии для немногочисленной когорты больных с исходным снижением CD4+ .

Введение. Болезнь Гоше (БГ) — заболевание из группы лизосомных болезней накопления. Заместительная ферментная терапия (ЗФТ) является современным стандартом лечения БГ. Поддерживающий режим ЗФТ до настоящего времени не разработан.
Цель исследования: разработка оптимального, научно-экономически обоснованного режима поддерживающей ЗФТ у взрослых больных БГ I типа.
Материалы и методы. В исследование включено 100 взрослых больных БГ I типа, достигших целей лечения на фоне как минимум двух лет ЗФТ в стандартном режиме. Больные переводились на поддерживающий режим ЗФТ, заключавшийся в увеличении интервала между инфузиями рекомбинантного фермента до 4 недель, в дозе 15– 20 ед./кг массы тела. Оценка эффективности поддерживающего режима ЗФТ проводилась c интервалом 1 раз в 12 месяцев и включала основные показатели активности БГ. Срок наблюдения больных в рамках исследования варьировал от 12 до 36 месяцев. Результаты. При использовании поддерживающего режима ЗФТ у больных БГ I типа, достигших целей лечения на фоне стандартного начального режима ЗФТ, сохраняется стабильность ранее достигнутого лечебного эффекта по всем критериям эффективности: не выявлено клинически значимых различий в показателях гемоглобина и тромбоцитов, размерах селезенки и степени специфической инфильтрации костного мозга бедренных костей.
Заключение: увеличение до 4 недель интервалов между инфузиями рекомбинантной глюкоцереброзидазы на протяжении 12, 24 и 36 месяцев не привело к ухудшению лабораторных и инструментальных показателей, ассоциированных с активностью БГ.
ОБЗОРЫ ЛИТЕРАТУРЫ
Введение. В основе патогенеза приобретенной апластической анемии (АА) лежит иммуноопосредованное развитие костномозговой недостаточности. Отсутствие однозначных причин развития иммунной агрессии делает актуальными исследования, направленные на изучение генетических нарушений в оставшемся пуле гемопоэтических стволовых клеток, в кроветворной нише, а также механизмов срыва иммунологической толерантности.
Цель настоящего обзора литературы — описание наиболее актуальных маркеров, позволяющих охарактеризовать больных АА в зависимости от возможного ответа на ИСТ и сформировать группы риска развития рефрактерности и клональной эволюции.
Основные сведения. Вероятность общей выживаемости больных АА, которым проведена программная иммуносупрессивная терапия (ИСТ), сопоставима с результатами трансплантации аллогенных гемопоэтических стволовых клеток крови (алло-ТГСК) от родственного донора в первой линии терапии. Согласно современным отечественным и международным рекомендациям, выбор тактики лечения больных АА определяется возрастом больного и наличием HLA-идентичного сиблинга. Методом выбора лечения больных младше 40 лет является алло-ТГСК от родственного HLA-идентичного донора, но возможность проведения алло-ТГСК ограничена наличием донора. Несмотря на то что вероятность бессобытийной выживаемости при проведении ИСТ уступает результатам алло-ТГСК, для большинства больных АА ИСТ остается основным методом лечения. С целью минимизации неблагоприятных исходов необходимо учитывать наличие предикторов эффективности лечения и вероятность развития поздней клональной эволюции уже на этапе диагностики АА. Оценка и формирование групп риска больных позволит на этапе планирования выбрать оптимальный подход, включающий добавление к ИСТ агонистов тромбопоэтиновых рецепторов, или поиск неродственного HLA-совместимого донора и переход к алло-ТГСК в более ранние сроки.
КЛИНИЧЕСКИЕ НАБЛЮДЕНИЯ

Введение. Подкожная панникулитоподобная Т-клеточная лимфома (ППТКЛ) относится к редкой группе кожных лимфопролиферативных заболеваний с клиническими проявлениями, напоминающими панникулит, α/β-цитотоксическим иммунофенотипом опухолевых клеток и разнонаправленным течением: от индолентных до агрессивных форм.
Цель работы — описать больных ППТКЛ с агрессивным клиническим течением заболевания и рефрактерностью к нескольким линиям химиотерапии.
Результаты. Представлены два клинических наблюдения больных с генерализованным характером поражения и наличием факторов неблагоприятного прогноза, у которых достигнуты полные продолжительные ремиссии заболевания в результате применения курсов химиотерапии с включением гемцитабина.
Заключение: несмотря на то, что у обоих больных ППТКЛ наблюдалась рефрактерность как минимум к трем видам лечения, использование гемцитабина позволило достигнуть длительных полных ремиссий заболевания.
Введение. Множественная миелома (ММ) является лимфопролиферативным заболеванием, длительность ремиссии которого сложно прогнозировать.
Цель работы: проанализировать молекулярно-генетический статус опухоли у больного с коротким периодом ремиссии в дебюте и рецидиве ММ и сопоставить с клиническим течением заболевания.
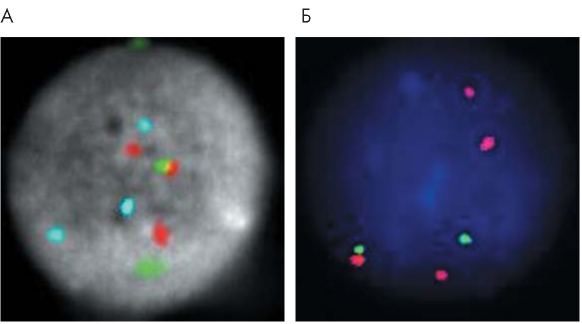
Материалы и методы. Соматические мутации определяли методом секвенирования по Сэнгеру. Уровень экспрессии генов анализировали с помощью секвенирования РНК на платформе Illumina. Для изучения хромосомных перестроек проводили флуоресцентную гибридизацию in situ (FISH-исследование).
Результаты. До начала лечения и в рецидиве заболевания у больного выявлена гетерозиготная клональная мутация с.182A>C (p.Q61P) в гене N-RAS, нарушающая регуляцию сигнального пути МАРК. Транскриптомный анализ, выполненный методом RNA-seq, показал резкое усиление экспрессии гена IL6 при рецидиве (в 30 раз), которое могло послужить пусковым механизмом прогрессии множественной миеломы, поскольку этот цитокин стимулирует клеточную пролиферацию, активируя различные сигнальные пути (MAPK, JAK-STAT, PI3K). Прогрессия заболевания сопровождалась также усилением экспрессии ключевых регуляторных генов (с-MYC, Notch2, MDM, RAF1, STAT4, mTOR) и резким уменьшением экспрессии генов иммуноглобулинов, вызвавшим у больного глубокий иммунодефицит. При молекулярно-цитогенетическом исследовании (FISH) в дебюте заболевания была выявлена трисомия по хромосомам 5, 9, и 15. Рецидив заболевания сопровождался амплификацией локуса 1q21 при сохранении гипердиплоидии.
Заключение. Для прогноза длительности периода ремиссии необходимо проводить комплексный молекулярногенетический скрининг.

ISSN 2411-3042 (Online)





































