


Содержание
Перейти к:
https://doi.org/10.35754/0234-5730-2020-65-2-190-207
Перейти к:
Введение. Болезни тяжелых цепей (БТЦ) представляют собой редкие В-клеточные лимфопролиферативные заболевания, не имеющие классической клинической картины. Характерной особенностью является секреция фрагментов тяжелых цепей различных изотипов иммуноглобулинов. В настоящее время известно четыре варианта заболеваний: μ, γ, α и δ.
Цель. Описание клинического наблюдения БТЦ-μ, скрывавшейся под маской системного амилоидоза, и связанные с этим трудности первичной диагностики.
Основные сведения. Представлен редкий клинический случай БТЦ-μ в сочетании с системным амилоидозом (амилоидоз легких цепей (light chain amyloidosis — AL) и транстиретиновый амилоидоз (transthyretin amyloidosis — ATTR) и неамилоидными депозитами у больного 64 лет. Тяжесть состояния была обусловлена хронической сердечной недостаточностью, полинейропатией. При обследовании был установлен диагноз «Макроглобулинемия Вальденстрема». Диагностировать амилоидоз не удалось. Проводилась иммунохимиотерапия по программе RB (ритуксимаб и бендамустин). Эффект терапии был минимальным и кратковременным. Состояние больного прогрессивно ухудшалось, и он умер вследствие острой сердечно-сосудистой недостаточности. Основной диагноз был пересмотрен в пользу БТЦ-μ. При аутопсии было обнаружено распространенное амилоидное и неамилоидное поражение органов и тканей.
Конфликт интересов: Авторы заявляют об отсутствии конфликта интересов.
Финансирование. Исследование не имело спонсорской поддержки.
Охота В.К., Рыжко В.В., Ковригина А.М., Шуплецова И.А., Соболева Н.П., Грибанова Е.О. Болезнь тяжелых цепей-μ в сочетании с системным амилоидозом и неамилоидными депозитами. Трудности диагностики и терапии. Гематология и трансфузиология. 2020;65(2):190-207. https://doi.org/10.35754/0234-5730-2020-65-2-190-207
Okhota V.K., Ryzhko V.V., Kovrigina A.M., Shupletsova I.A., Soboleva N.P., Gribanova E.O. μ-Heavy chain disease associated with systemic amyloidosis and non-amyloid deposits. Diffi culties in diagnosis and therapy. Russian journal of hematology and transfusiology. 2020;65(2):190-207. (In Russ.) https://doi.org/10.35754/0234-5730-2020-65-2-190-207
Болезни тяжелых цепей (БТЦ) — это редкие клональные B-клеточные заболевания из группы парапротеинемических гемобластозов, характерной особенностью которых является присутствие в сыворотке крови и/или моче структурно дефектных H-цепей иммуноглобулинов, лишенных большей части Fd- фрагмента, т. е. части H-цепи, входящей в состав Fab-фрагмента молекулы иммуноглобулина [1, 2]. Клиническое течение болезней тяжелых цепей варьирует от асимптома- тического до крайне агрессивного. Прогностические факторы риска до настоящего времени не определены, стандартная терапия не разработана [3].
Молекулы нормальных иммуноглобулинов состоят из двух типов полипептидных цепей — тяжелой (H — heavy) и легкой (L — light). Мономерный иммуноглобулин содержит две Н- и две L-цепи. Каждая из тяжелых и легких цепей имеет в своем составе константные (С — constant) и вариабельные (V — variable) домены. Выделяют 5 основных изотипов Н-цепей — μ, γ, α, δ и ε, и в зависимости от их структуры существует 5 классов молекул иммуноглобулинов (Ig) — IgM, IgG, IgA, IgD и IgE (латинские буквы в названии иммуноглобулинов соответствуют греческим в обозначении изотипов Н-цепей). Существует два типа легких цепей — К и λ [4].
Синтез иммуноглобулинов значительно отличается от синтеза других белков. Каждая тяжелая цепь иммуноглобулина кодируется четырьмя генами, которые находятся на 14-й хромосоме. Каждая легкая цепь кодируется группой из трех различных генов. Гены легкой цепи κ расположены на 2-й хромосоме, гены легкой цепи λ — на 22-й. Вариабельные (V) и константные (С) области молекул иммуноглобулинов кодируются разными группами генов. В процессе синтеза иммуноглобулинов после окончания трансляции и посттрансляционного процессинга тяжелые и легкие цепи взаимодействуют между собой и образуют нековалентные связи, в результате чего происходит сборка четырехцепочечной молекулы, содержащей две тяжелые (H) и две легкие (L) цепи [4].
В зависимости от класса иммуноглобулина (G, A, M и D) существует четыре варианта заболевания: α, γ, μ и δ. БТЦ IgE не описана, однако нельзя исключить ее существования. Клинически и морфологически все варианты заболевания различны между собой, но схожи продукцией тяжелой цепи, не способной связывать легкие К и λ цепи [1, 5]. В таблице 1 представлены основные иммунофенотипические характеристики опухолевых клеток при разных вариантах БТЦ.
Таблица 1. Иммунофенотипические характеристики опухолевых клеток при разных вариантах БТЦ [6]
Table 1. Immunophenotypic characteristics of tumor cells in different variants of heavy chain disease [6]
| Тип тяжелой цепи Type of heavy chain | Фенотип опухолевых клеток Tumor cell phenotype |
|---|---|---|
БТЦ-а а-heavy chain disease | IgA Immunoglobulin A | Пан-В-клеточные антигены+ Pan-B-cell antigens CD138+ CD5- CD10- CD20- |
БТЦ-γ γ-heavy chain disease | IgG Immunoglobulin G | CD19+ CD20+ Mum1/IRF4+CD38+ CD138+ CD5- CD10- |
БТЦ-μ μ-heavy chain disease | IgM Immunoglobulin M | CD19+ CD20+ CD38+ kappa-light chain + (редко) CD5+ (редко) |
Примечание. Kappa-light chain — свободная легкая цепь-каппа; Muml/IRF4 — Multiple myeloma 1/Interferon Regulatory Factor 4 protein; CD — clusters of differentiation.
БТЦ-а (синоним — иммунопролиферативное заболевание тонкой кишки, IPSID — immunoproliferative small intestinal disease) — вариант MALT-лимфомы (mucosa-associated lymphoid tissue) [1]. БТЦ-α чаще называют cредиземноморской лимфомой, которая впервые была описана в 1968 г. [7], с тех пор в литературе упоминается более 400 случаев [1]. Заболевание подразделяют на три формы в зависимости от локализации поражения: гастроинтестинальную, респираторную и лимфоидную. В подавляющем большинстве случаев встречается именно гастроинтестинальная форма, более известная как иммунопролиферативное заболевание тонкого кишечника. В таких случаях в собственной пластинке слизистой оболочки тонкой кишки отмечается выраженный плазмоклеточный инфильтрат с примесью лимфоцитов, могут присутствовать лимфоэпителиальные поражения, характерные для MALT-лимфомы. Возможна трансформация в диффузную В-крупноклеточную лимфому [8]. Костный мозг при БТЦ-α, как правило, в процесс не вовлечен [9].
Болезнь поражает преимущественно детей и молодых людей в возрасте от 10 до 35 лет из группы низкого социального статуса. Клиническая картина характеризуется потерей веса, диареей, длительными болями в животе, мальабсорбцией, а в тяжелых случаях — кишечной непроходимостью и асцитом [10].
БТЦ-γ, или болезнь Франклина, получила название от имени впервые описавшего ее в 1964 г. врача [11]. Всего известно около 130 случаев заболевания [1]. В равной степени встречается у мужчин и женщин, средний возраст заболевших 68 лет [12—14]. Классические проявления БТЦ-γ включают генерализованную лимфаденопатию (34% случаев), спленомегалию (30%) и вовлечение костного мозга (30%). Клиническая картина чаще всего напоминает лимфоплазмоцитарную лимфому, реже — лимфому Ходжкина и неходжкинские лимфомы. Часть описанных случаев ассоциирована с аутоиммунными заболеваниями, такими как ревматоидный артрит, аутоиммунная гемолитическая анемия и идиопатическая тромбоцитопеническая пурпура [15, 16]. Морфологическая картина гетерогенна, некоторые случаи напоминают лимфому из клеток маргинальной зоны. Наиболее часто в лимфатических узлах выявляют полиморфную инфильтрацию лимфоцитами, плазмоцитоидными лимфоцитами, плазматическими клетками, иммунобластами, гистиоцитами и эозинофилами, что может напоминать лимфому Ходжкина или ангиоиммунобластную Т-клеточную лимфому. В других случаях преобладают плазматические клетки, что напоминает плазмоцитому. При иммунофенотипировании, как правило, не определяют экспрессию легких цепей [17].
Jan Van Keer и соавт. [18] в 2016 г. описали два клинических случая БТЦ-α и БТЦ-γ, которые протекали без признаков опухолевой пролиферации, в результате чего авторы предположили возможность существования БТЦ, протекающей как моноклональная гаммапатия неясного значения.
БТЦ-μ является одним из редчайших вариантов болезни тяжелых цепей, в настоящее время известно около 40 случаев. В 1969 и 1970 гг. F. A. Forte и H. S. Ballard [19, 20] независимо друг от друга впервые описали два случая БТЦ-μ. Оба больных были мужчинами, возраст их был 58 и 59 лет, у обоих были выраженные боли в суставах.
При БТЦ-μ в центральной части N-концевого участка μ-цепи иммуноглобулина происходит разрыв. В результате молекула иммуноглобулина теряет часть вариабельного домена (VH) и большую часть первого константного домена (GH1), которые способны присоединять соответствующую легкую цепь. В нормальных условиях при отсутствии ассоциированной легкой цепи тяжелая цепь иммуноглобулина подвергается протеасомной деградации [21]. Поскольку разрыв в области N-концевого участка μ-цепи иммуноглобулина приводит к утрате участка, обеспечивающего связь с протеасомой, деградация тяжелой цепи становится невозможной [22]. Таким образом, патологическая μ-цепь секретируется и циркулирует в крови. Синтез легких цепей при этом не нарушен, одновременно происходит секреция моноклональных свободных легких цепей (белок Бенс-Джонса). В результате чего при БТЦ-μ можно обнаружить белок Бенс-Джонса.
Большинство заболевших — это белые мужчины, медиана возраста которых — 58 лет. Этиология и заболеваемость БТЦ-μ неизвестны [12]. Заболевание клинически и морфологически схоже с хроническим лимфоцитарным лейкозом/лимфомой из малых лимфоцитов. Вместе с тем, описаны случаи, протекавшие с клинической картиной диффузной В-крупноклеточной лимфомы, лимфоплазмоцитарной лимфомы, множественной миеломы, макроглобули- немии Вальденстрема [1, 3]. Встречаются единичные описания БТЦ-μ в сочетании с системной красной волчанкой [23]. Как правило, в костном мозге обнаруживают плазматические клетки с вакуолизированной цитоплазмой и примесь мелких округлых лимфоцитов [3, 5]. Пик моноклонального белка на электрофореграмме наблюдают в половине случаев. У большей части больных выявляется протеинурия Бенс-Джонса, преимущественно k-типа, экскреция легкой цепи λ встречается реже [24]. Почти у всех больных имеется спленомегалия, а гепатомегалия выявляется у трети больных. У 40% больных заболевание протекает с периферической лимфаденопатией [1, 12].
БТЦ-μ в разные годы диагностировали в США, Японии, Италии, Германии, Великобритании и Кот- д'Ивуаре [23, 25—29]. J. Bonhomme и соавт. в 1974 г. описали 45-летнего больного с симптомами опухолевой интоксикации, анемией, внутрибрюшной лимфаденопатией, спленомегалией, у которого в костном мозге была выявлена лимфоплазмоцитарная инфильтрация, а в сыворотке и моче обнаружена μ-тяжелая цепь [25].
M. Witzens и соавт. [26] наблюдали 66-летнего больного с БТЦ-μ, гиперглобулинемией и положительной пробой Кумбса. Заболевание протекало с глубокой анемией, умеренной спленомегалией при отсутствии лимфаденопатии. В костном мозге была картина миелодисплазии.
Разнообразие представленных случаев свидетельствует об отсутствии классической клинической картины заболевания, которое может протекать наподобие различных гематологических опухолей или быть с ними ассоциированным, нередко является случайной находкой.
Известно о трех случаях несекретирующей формы БТЦ-μ, когда диагноз был установлен на основании обнаружения μ-цепи в цитоплазме плазматических клеток с помощью реакции иммунофлуоресценции [24, 30, 31].
Есть наблюдения БТЦ-μ, протекавшей как моноклональная гаммапатия. D. L. Wahner-Roedler и соавт. [27] описали больного БТЦ-μ, которого наблюдали в течение 4 лет до появления панцитопении, гепатоспленомегалии и забрюшинной лимфаденопатии. В результате лечения циклофосфамидом, винкристином и преднизолоном удалось достичь улучшения состояния, однако через год констатировали трансформацию опухоли в крупноклеточную лимфому.
В лечении БТЦ-μ в разное время применялись режимы химиотерапии с включением в схемы антрациклиновых и алкилирующих агентов в сочетании с глюкокортикостероидными гормонами. A. Maeda и соавт. [32] описали 5-летнее наблюдение за больной БТЦ-μ, получавшей химиотерапию флудараби- ном, затем циклофосфамидом в течение 2 лет. Позже у нее обнаружили опухоль правой молочной железы. Гистологическая картина биоптата была характерной для диффузной В-крупноклеточной лимфо- мы, а при иммуногистохимическом исследовании клетки опухоли были позитивны на цитоплазматическую μ-цепь.
БТЦ-δ является самой редкой патологией. Описан единственный случай, когда у больной была диагностирована множественная миелома на основании плазмоклеточной инфильтрации костного мозга и остеолитических очагов в костях черепа, но при иммунохимическом исследовании в сыворотке крови обнаружили δ-тяжелую цепь, не ассоциированную ни с К-, ни с λ-легкими цепями. Диагноз был изменен на БТЦ-δ, протекавшей с клинической картиной множественной миеломы [33]. Краткая сравнительная характеристика БТЦ представлена в таблице 2.
Таблица 2. Клинические характеристики вариантов болезни тяжелых цепей [5]
Table 2. Clinical characteristics of different types of heavy chain disease [5]

Примечание. ХЛЛ — хронический лимфоцитарный лейкоз.
Note. CLL — chronic lymphocytic leukemia.
Нами описан клинический случай БТЦ-μ со следовой моноклональной секрецией IgM, экскрецией легкой цепи λ, лимфоплазмоцитарной инфильтрацией костного мозга, мутацией в гене MYD88+, системным амилоидозом и неамилоидными депозитами в органах и тканях.
Целью работы является описание клинического наблюдения БТЦ-μ, скрывавшейся под маской системного амилоидоза, и связанные с этим трудности первичной диагностики.
Больной П., 64 лет. В апреле 2017 г. у него впервые возникли боли в голеностопных и коленных суставах, парестезии кистей, стоп и голеней по типу «перчаток» и «гольф». В июне того же года появились отеки нижних конечностей, асцит и одышка при умеренной физической нагрузке. Зимой 2018 г. был дважды госпитализирован в стационары г. Москвы и Московской области, где были выявлены периферическая лимфаденопатия, анасарка (двусторонний плевральный выпот и асцит). Выполнены плевроцентез и лапароцентез. Клетки плеврального выпота были представлены преимущественно лимфоцитами. Проточная цитометрия для определения фенотипа лимфоцитов не проводилась. При гистологическом исследовании биоптата шейного лимфатического узла рисунок строения узла был стерт, отмечался склероз капсулы, атрофия и липоматоз медуллярного слоя. Изменения были расценены как реактивный лимфаденит. Методами инструментального обследования органов грудной клетки, брюшной полости, желудочно-кишечного тракта, мочеполовой и эндокринной систем были исключены онкологические заболевания. Результаты исследования альфа-фетопротеина, простатспецифического антигена, раково-эмбрионального антигена были отрицательными. Содержание ревматоидного фактора было в пределах нормы. При электронейромиографии было обнаружено грубое аксонально-демиелинизирующее поражение чувствительных и двигательных периферических нервных волокон верхних и нижних конечностей. Больной на протяжении 30 лет страдал циклотимией, по поводу чего постоянно получал терапию, в том числе карбамазепином. Был выписан с диагнозом: «Лекарственная болезнь».
В марте 2018 г. поступил в ФГБУ «НМИЦ гематологии» Минздрава России. При поступлении состояние было тяжелым, предъявлял жалобы на одышку, онемение кистей и голеней, стоп. При осмотре были обнаружены увеличенные подчелюстные лимфатические узлы, васкулитоподобные высыпания на коже предплечий, плеч и живота, отеки нижних конечностей до середины бедер. При аускультации легких с обеих сторон на фоне ослабленного дыхания выслушивались незвучные влажные хрипы. Перкуторно определялось двустороннее притупление легочного звука ниже угла лопатки. Отмечалась ортостатическая гипотония — АД 100/70 мм рт. ст. в положении лежа и 80/60 мм рт. ст. в положении сидя. Сердечные тоны были приглушены, ритм был правильным, частота сердечных сокращений — 90 уд./мин. Ж.ивот увеличен в размерах за счет асцита. Суточный диурез составлял 800—900 мл.
В гемограмме определяли: нормальную концентрацию гемоглобина — 124 г/л, незначительное увеличение количества тромбоцитов 369 х 10 9/л, лейкоциты 8,2 х 10 9/л, нормальную лейкоцитарную формулу и увеличение СОЭ до 33 мм/ч. В биохимическом анализе крови общий белок, альбумин, глобулин, печеночные трансаминазы были в пределах нормы. Концентрация азота мочевины была 4,6 ммоль/л, повышены активности лактатдегидрогеназы — 522 ед/л, щелочной фосфатазы 136 ед/л и гамма-глютамилтранспептидазы — 55,4 ед/л. В коагулограм- ме активированное частичное тромбопластиновое время — 28,2 с, протромбин по Квику — 87 %, активность антитромбина III — 113 %, агрегация тромбоцитов с АДФ — 73%, гиперфибриногенемия 6,6 г/л. Относительная плотность мочи была выше нормы — 1,035, протеинурия составляла 1,0 г/л, проба на уробилиноген была положительной. Обращало на себя внимание увеличение концентраций сердечных маркеров: тропонина и NT-proBNP — 0,18 мг/мл и 10 734 пг/мл соответственно.
На электрокардиограмме были зарегистрированы: низкая амплитуда в стандартных отведениях, признаки гипертрофии левого предсердия (митральный зубец P), атриовентрикулярная блокада I степени. При эхокардиографии визуализировались гидроперикард, дилатация обоих предсердий, правого желудочка и легочной артерии, толщина межжелудочковой перегородки — 23 мм (норма — до 12 мм), толщина стенки левого желудочка — 22 мм (норма — до 11 мм), фракция выброса левого желудочка составила 31%, систолическое давление в легочной артерии — 35 мм рт. ст. Обнаруженные изменения свидетельствовали о рестриктивной кардиомиопатии, протекавшей с клинической картиной сердечной недостаточности. Внутригрудная, внутрибрюшная и забрюшинная лимфаденопатия, двусторонний гидроторакс, гепатомегалия были подтверждены результатами компьютерной томографии и ультразвукового исследования. Размеры селезенки были 120 х 50 мм.
В сыворотке крови на фоне гипогаммаглобулинемии были выявлены следовая секреция моноклонального иммуноглобулина МХ, увеличение количества СЛЦ-λ до 868 мг/л. Экскреция белка Бенс-Джонса λ-типа составляла 0,24 г/л, суммарная экскреция — 0,1 г при суточном диурезе 0,41 л. Концентрация β2-микроглобулина была увеличена до 4,45 мг/л.
В костном мозге были плазматические клетки с вакуолизированной цитоплазмой (рис. 1), умеренная очагово-диффузная инфильтрация, представленная мелкими лимфоидными клетками, клетками с лимфоплазмоцитоидной морфологией и зрелыми плазматическими клетками (рис. 2 А и Б). Иммунофенотип клеток лимфоплазмоцитарной инфильтрации отображен на рисунке 3. Характеристики примененных в исследовании антител указаны в таблице 3. Лимфоциты были представлены фенотипом: CD19+CD20+CD22+CD81+CD200+CD79b- CD5-CD23-k/X-, плазматические клетки CD45+CD 38+CD138+CD19+CD56-CD27+CD81+CD117-cytX+. Обнаружена мутация p. L625P в гене MYD88.
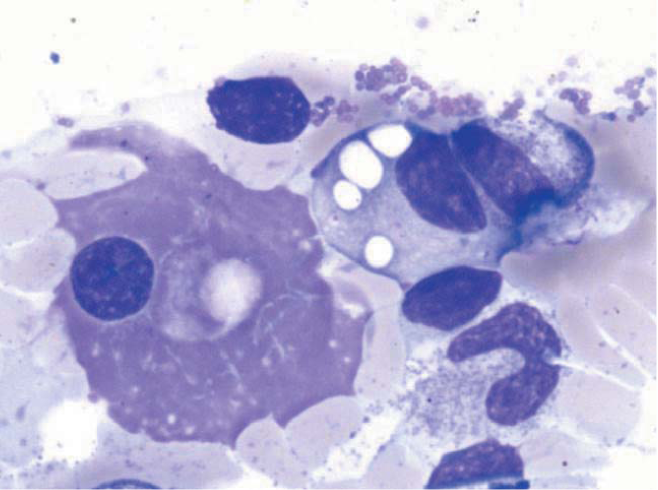
Рисунок 1. Цитологический препарат костного мозга. Плазматическая клетка с вакуолизированной цитоплазмой. Окраска по Паппенгейму. Увеличение ×1000
Figure 1. Bone marrow aspirate shows vacuolated plasma cell. Pappenheim stain. Magnification ×100
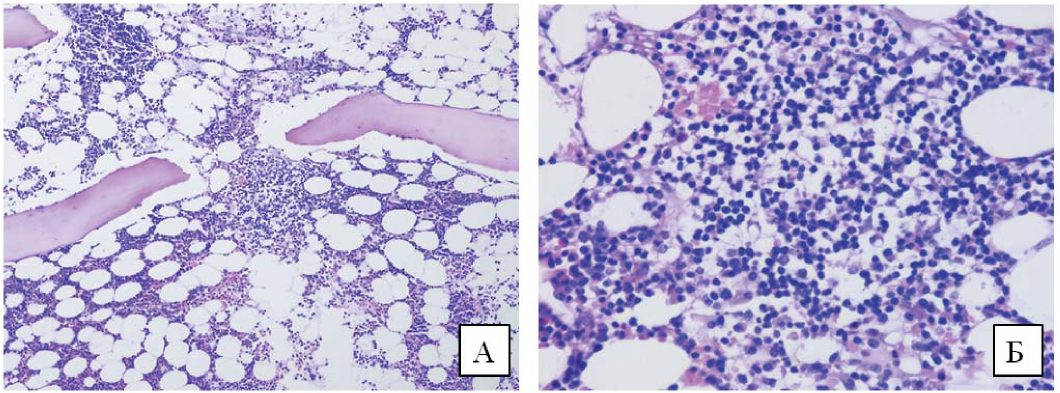
Рисунок 2. Трепанобиоптат костного мозга. Очагово-интерстициальная лимфоплазмоцитарная инфильтрация. Окраска гематоксилином и эозином. А. Увеличение х100. Б. Увеличение х400
Figure 2. Bone marrow trephine biopsy shows focal-interstitial lymphoplasmacytic infiltration. Hematoxylin and eosin staining.A. Magnification ×100.

Рисунок 3. Трепанобиоптат костного мозга. Иммуноферментный метод. Увеличение х200. Экспрессия клетками очагово-интерстициальной лимфоплазмоцитарной инфильтрации CD20 (А), CD19 (Б). В зрелых плазматических клетках и клетках с лимфоплазмоцитоидной морфологией отмечается экспрессия CD138 (В), IgM (Г), рестрикция lambda-цепи (Д); kappa-позитивные клетки единичны (Е)
Figure 3. Bone marrow trephine biopsy. Enzyme-linked immunosorbent assay. Magnification х200. Cells of focal-interstitial lymphoplasmacytic infiltration express CD20 (А) and CD19 (Б). Mature plasma cells with lymphoplasmocytoid morphology express CDtSS (В) and IgM (Г). There are lambda-chain restriction (Д) and single kappa-positive cells (Е)
Таблица 3. Характеристики антител, примененных в исследовании
Table 3. Characteristics of antibodies used in the study
Антитело Antibody | Производитель, страна Manufacturer, Country | Клон Clone |
|---|---|---|
CD20 | Dako, Дания Dako, Denmark | L-26 |
CD3 | Leica, Великобритания Leica, Great Britain | NCL- L-CD3-565 |
CD10 | Leica, Великобритания Leica, Great Britain | 4B12 |
CD19 | Leica, Великобритания Leica, Great Britain | LE-CD19 |
CD56 | Dako, Дания Dako, Denmark | 123C3 |
CD138 | Epitomics, США Epitomics, USA | EP201 |
VS38c | Dako, Дания Dako, Denmark | VS38c |
IgM | Dako, Дания Dako, Denmark | R1/69 |
IgG | Dako, Дания Dako, Denmark | Polyclonal |
IgA | Dako, Дания Dako, Denmark | Polyclonal |
Cyclin D1 | Cell Marque, США Cell Marque, USA | SP4 |
Kappa | Dako, Дания Dako, Denmark | Polyclonal |
Lambda | Dako, Дания Dako, Denmark | Polyclonal |
Примечание. CD — кластеры дифференцировки, kappa — каппа легкая цепь, lambda — лямбда легкая цепь.
Note. CD — clusters of differentiation, kappa — kappa light chain, lambda — lambda light chain.
Плевральная жидкость содержала лимфоциты с иммунофенотипом CD20+CD25+CD45+CD81+CD5-CD1 0-CD38-CD103-.
Таким образом, было подтверждено наличие у больного зрелой лимфоплазмоцитарной опухоли с вовлечением в процесс костного мозга и плевры.
Дифференциальный диагноз проводили между POEMS-синдромом (Polyneuropathy, Organomegaly, Endocrinopathy, М-protein, Skin Changes — полинейропатия, органомегалия, эндокринопатия, парапротеин, кожные изменения), системным AL-амилоидозом, лимфоплазмоцитарной лимфомой и макроглобулин- емией Вальденстрема.
POEMS-синдром включает в себя большие диагностические критерии: полинейропатию, моноклональную секрецию, склеротические изменения костной ткани, гистологическую картину болезни Кастлемана в лимфатическом узле и высокий уровень фактора роста эндотелия сосудов (Vascular Endothelial Growth Factor — VEGF). Малыми критериями являются органомегалия, отечный синдром, не обусловленный сердечной недостаточностью или нефротическим синдромом, эндокринопатия, кожные изменения, отек зрительного нерва и тромбозы. Диагноз является достоверным при наличии не менее трех больших критериев и одного малого [34]. В настоящем наблюдении POEMS-синдром был заподозрен на основании полинейропатии (онемение и парестезии кистей, стоп и голеней по типу «перчаток» и «гольф», доказанное аксонально-демиелинизирующее поражение нервных волокон), секреции парапротеина в сыворотке крови и экскреции его с мочой. Однако POEMS-синдром был исключен из-за нехарактерной для POEMS-синдрома лимфоплазмоцитарной инфильтрации костного мозга и наличия только двух из трех обязательных критериев, а именно парапротеина в сыворотке и периферической полинейропатии.
Амилоидоз при IgM ассоциированных заболеваниях встречается менее чем в 5% случаев [35, 36]. Тем не менее, учитывая васкулитоподобные высыпания на коже, полинейропатию, ортостатическую гипотонию, рестриктивную кардиомиопатию, с целью исключения амилоидоза были выполнены биопсии слизистой десны, желудка и подкожно-жировой клетчатки и костного мозга для гистохимического окрашивания Конго красным. При просмотре в поляризованном свете ни в одном из образцов отложений амилоида выявлено не было.
Кроме гипервискозного синдрома и анемии симптомами макроглобулинемии Вальденстрема могут быть полинейропатия и амилоидоз [37].
На основании лимфоплазмоцитарной инфильтрации костного мозга, следовой секреции парапротеина Mλ (PIgMX), протеинеурии Бенс-Джонса λ, наличия мутации p. L625P в гене MYD88 был установлен диагноз: «Макроглобулинемия Вальденстрема, протекающая с периферической полинейропатией, специфическим поражением плевры, рестриктивной миокардиопатией, сердечной недостаточностью, гепатомегалией».
Учитывая вариант лимфопролиферативного заболевания и тяжелый соматический статус больного, было решено провести противоопухолевую иммунохимиотерапию в режиме RB (ритуксимаб 375 мг/м2 и бендамустин 90 мг/м2). Выбранная программа позволяла сбалансировать эффективность лечения и химиотерапевтическую токсичность [38].
В первую неделю после окончания иммунохимиотерапии наблюдалось улучшение состояния, побледнели высыпания на коже, уменьшились отеки и размеры печени, увеличился диурез, исчезла внутрибрюшная лимфаденопатия.
Достигнутый эффект был кратковременным. В течение последующих нескольких дней усилилась одышка, возникли приступы сердечной астмы, увеличился двусторонний гидроторакс, появились новые васкулитоподобные высыпания в шейно-воротниковой зоне, усугубилась полинейропатия. Иммунохимический ответ был минимальным: сохранялась следовая секреция PIgMX и протеинурия Бенс-Джонса λ- 0,06 г/сут. Однако в сыворотке крови методом иммунофиксации были обнаружены свободные моноклональные тяжелые цепи μ-класса. Диагноз был изменен на БТЦ-μ (рис. 4 и 5). Несмотря на проведенное специфическое лечение, комплексную сопроводительную терапию, состояние больного стремительно ухудшалось, и он умер от острой сердечно-сосудистой недостаточности в апреле 2018 г. Таким образом, от момента госпитализации до летального исхода прошло 6 недель.

Рисунок 4. Иммунофиксация белков сыворотки крови при малом разведении. Выявлена следовая секреция PIg МХ и белок тяжелых цепей μ (красные стрелки)
Figure 4. Serum proteins immunofixation at low dilution. Secretion of monoclonal Ig Μλ and μ heavy chain were revealed (red arrows)

Рисунок 5. Иммунофиксация белков мочи. На фоне клубочковой протеинурии подтверждено наличие белка Бенс-Джонса λ-типа
Figure 5. Urine proteins immunofixation. Bens-Jones protein λ-type were revealed (red arrows)
При исследовании аутопсийного материала обнаружен двусторонний гидроторакс (600 мл выпота в правой плевральной полости и 1000 мл в левой). Наблюдалось уплотнение и воскообразная консистенция большого сальника (рис. 6) и забрюшинной клетчатки с потерей обычной макроскопической структуры. Обнаружена эксцентрическая гипертрофия миокарда (масса сердца 670 г, толщина стенки левого желудочка 2,5 см, правого 0,8 см), с воскообразными белесоватыми отложениями на эпикарде. Селезенка была со множественными мелкими включениями округлой формы до 0,3 см в диаметре, напоминающими зерна саго, масса селезенки была 340 г. Другие органы макроскопически были без особенностей. При микроскопическом исследовании обнаружены аморфные эозинофильные депозиты в стенках сосудов легких, печени, почек, селезенки, отдельных сосудах костного мозга, а также в строме миокарда, щитовидной железы, простаты, клубочках почки, периартериолярных зонах селезенки, массивные белковые отложения в паранефральной клетчатке, эпикарде. Все образцы были окрашены Конго красным и исследованы в поляризованном свете.
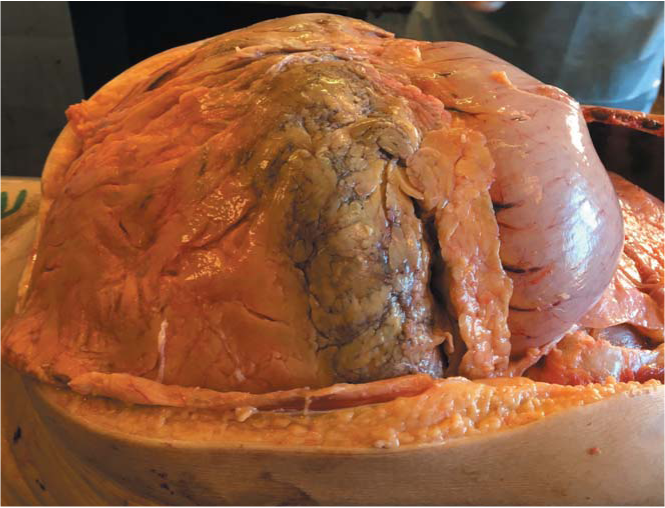
Рисунок 6. Большой сальник с потерей нормальной макроскопической структуры, воскообразной консистенции. Аутопсийный материал
Figure 6. Large omentum with waxy consistency and loss of normal macroscopic structure. Autopsy material
В образцах, за исключением миокарда, простаты и селезенки, определялись аморфные конгофильные депозиты с эффектом двойного лучепреломления в поляризованном свете, что свидетельствовало о наличии амилоида (рис. 7). Аморфные эозинофильные депозиты в миокарде, простате и периартериолярных зонах селезенки (рис. 8) только очагово демонстрировали зеленовато-яблочное свечение в поляризованном свете. Остальная масса белковых депозитов не демонстрировала конгофилию, что указывало на ее неамилоидную природу. Для установления природы амилоидного парапротеина было проведено иммуногистохимическое исследование с антителами к P-компоненту, L-цепям Ig, транстиретину, АА, IgM. В стенках сосудов аморфные массы окрасились при реакции с анти-P компонентом, IgM и транстире- тином. Ввиду выраженных явлений абсорбции белковых масс к иммуноглобулинам для более точного определения белка-предшественника амилоида ткань миокарда была исследована с помощью конфокальной микроскопии (ПСПбГМУ им. акад. И.П. Павлова, Санкт-Петербург). В ткани миокарда субэндотели- ально и в строме были выявлены депозиты амилоида сочетанной природы: AL, ATTR (белки-предшественники — l-цепь и транстиретин) (рис. 9 и 10).
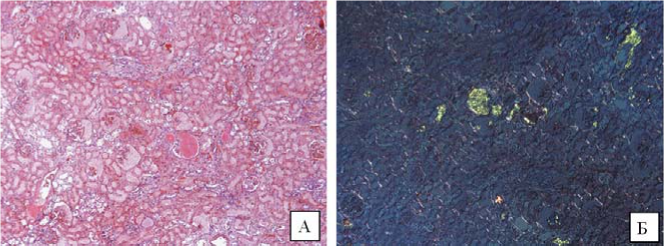
Рисунок 7. Аутопсийный материал ткани почки. А. Конгофильные депозиты в отдельных клубочках и стенке сосудов мелкого калибра. Гистохимическая окраска Конго красным. Увеличение *50. Б. Эффект двойного лучепреломления в конгофильных депозитах. Поляризованный свет. Увеличение *50
Figure 7. Autopsy examination of kidney tissue. A. Congophilic deposits in single glomeruli and the walls of small-caliber vessels. Histochemical staining of Congo red. Magnification *50. B. Birefringence effect in congophilic deposits. Polarized light. Magnification. *50

Рисунок 8. Отложения амилоида в стенке центральной артерии селезенки (конгофильные, с эффектом двойного лучепреломления в поляризованном свете) и массивные отложения парапротеина в периартериолярной зоне. Гистохимическое окрашивание Конго красным, поляризованный свет. Увеличение *50
Figure 8. Amyloid deposits in the wall of the central spleen artery (congophilic, with the birefringence effect in polarized light) and massive deposits of paraprotein in the periarteriolar zone. Histochemical staining of Congo red, polarized light. Magnification *50
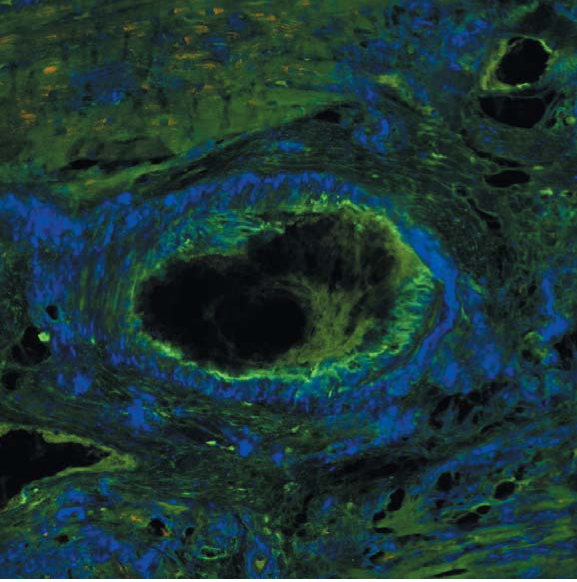
Рисунок 9. Изображение миокарда, полученное с помощью конфокальной микроскопии в ходе исследования с антителами к транстиретину (зеленый) и IgM (красный). Визуализируется отложение транстиретина под эндотелием, IgM — нет. Слайд любезно предоставлен сотрудниками кафедры патологической анатомии Первого СПбГМУ им. акад. И. П. Павлова д. м.н., проф. Байковым В. В. и к. м.н. Кузнецовой И. А.
Figure 9. Image of the myocardium obtained by confocal microscopy with antibodies to prealbumin (green) and IgM (red). Transthyretin deposition under the endothelium is visualized but IgM is not. The slide was kindly provided by the staff of the Department of Pathological Anatomy of the First St. Petersburg State Medical University Academician I. P. Pavlova MD, prof. Baykov V. V. and candidate of medical sciences Kuznetsova I. A.
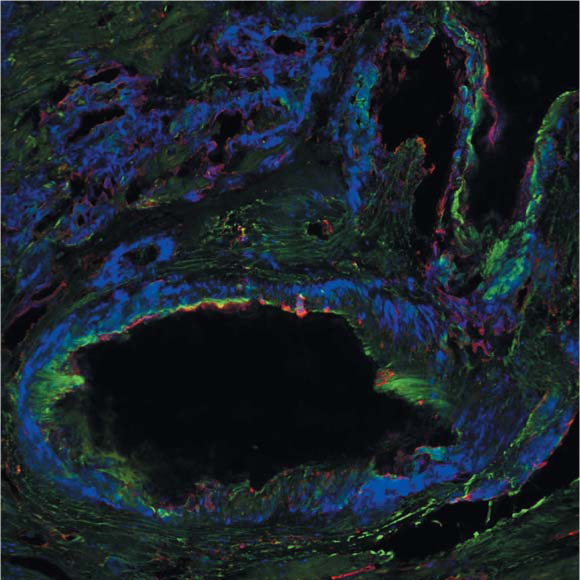
Рисунок 10. Изображение миокарда, полученное с помощью конфокальной микроскопии в ходе исследования с антителами к P-компоненту и lambda-цепи. Под эндотелием визуализируется отложение P-компонента (зеленый), lambda-цепи (красный) и их колокализация (желтый). Слайд любезно предоставлен сотрудниками кафедры патологической анатомии д. м.н., проф. Байковым В. В. и к. м.н. Кузнецовой И. А.
Figure 10. Image of the myocardium obtained by confocal microscopy with antibodies to the P-component and lambda-chain. Under the endothelium, deposition of the P component (green) is visualized, lambda chains (red) and their colocalization (yellow). The slide was kindly provided by the staff of the Department of Pathological Anatomy of the First St. Petersburg State Medical University Academician I. P. Pavlova MD, prof. Baykov V. V. and candidate of medical sciences Kuznetsova I. A.
При исследовании костного мозга тела грудного позвонка и верхней трети бедренной кости была выявлена интерстициально-очаговая лимфоплазмоцитарная инфильтрация с иммунофенотипом, идентичным инфильтрату в прижизненном трепанобиоптате костного мозга: CD20+, CD138+, IgM+, lambda+. В единичных сосудах были утолщены стенки, они давали положительную окраску на амилоид. В паранефральной клетчатке был обнаружен экстранодальный лимфоплазмоцитарный компонент с иммунофенотипом, аналогичным таковому в костном мозге (рис. 11 А и Б, рис. 12).
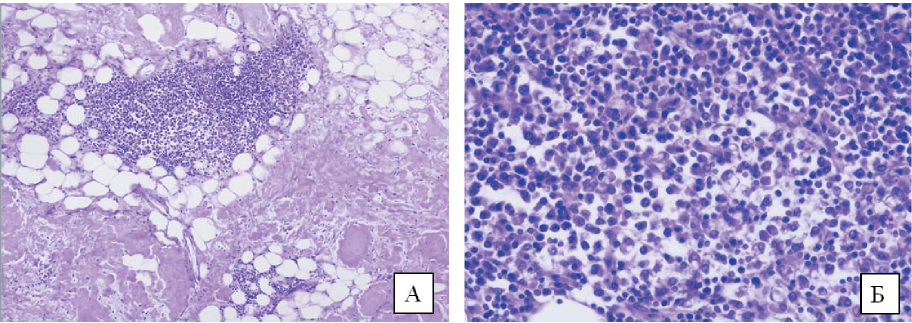
Рисунок 11. Лимфоплазмоцитарные скопления в паранефральной клетчатке. Аутопсийный материал. Окраска гематоксилином и эозином. А. Увеличение х100. Б. Увеличение х400
Figure 11. Lymphoplasmacyte infiltration in the paranephric tissue. Autopsy material. Hematoxylin and eosin staining. A. Magnification х 100. Б. Magnification х400

Рисунок 12. Лимфоплазмоцитарные скопления в паранефральной клетчатке. Аутопсийный материал. Иммуноферментный метод. Увеличение х200. В зрелых плазматических клетках и клетках с лимфоплазмоцитоидной морфологией отмечается экспрессия VS38c (А), IgM (Б), рестрикция lambda-цепи (В); kappa-позитивные клетки единичны (Г)
Figure 12. Lymphoplasmacyte infiltration in the paranephric tissue. Autopsy material. Enzyme-linked immunosorbent assay. Magnification х200. Mature plasma cells with lymphoplasmocytoid morphology express VS38c (A) and IgM (Б). There are lambda-chain restrictions (В) and single kappa-positive cells (Г)
БТЦ-μ — это крайне редкое заболевание, которое относят к группе парапротеинемических гемобластозов. Актуальным является вопрос происхождения опухолевой клетки при БТЦ. Фрагменты иммуноглобулинов являются продуктом аномального клеточного синтеза, а не постсинтетических изменений. Можно предположить, что ключевые перестройки происходят на уровне зрелого В-лимфоцита, прошедшего этап фолликулярной дифференцировки в герминальном центре лимфоидного фолликула [2]. Вместе с тем нельзя исключить вероятность поломок и на более ранних стадиях клеточного развития. D. Corcos и соавт. [39] в 2011 г. высказали предположение, что поврежденная тяжелая цепь, входящая в состав трансмембранного В-клеточного рецептора, может способствовать антиген-независимому формированию и передаче сигнала в клетку по типу хронической антигенной стимуляции, что потенциально может привести к клональной опухолевой пролиферации.
БТЦ не имеет характерной клинической картины, в результате чего диагностика представляет собой большие трудности. С момента первого описания в 1969 г. [19] прошло уже 50 лет, а известно в общей сложности около 40 случаев, поэтому каждое новое наблюдение представляет собой исключительную ценность с точки зрения клиники, диагностики и терапевтического подхода.
Трудности первичной иммунохимической диагностики, вероятно, были обусловлены малым количеством тяжелой μ-цепи в сыворотке. Для проведения реакции иммунофиксации и образования преципитата соотношение антигена (исследуемого белка) и антител (специфической антисыворотки) должно быть эквивалентным. В случае избытка антител или антигена преципитат растворяется, реакция иммунофиксации может быть ложноотрицательной. Разведение буферным раствором исследуемой сыворотки подбирается индивидуально в зависимости от количества иммуноглобулина. При проведении первичной иммунофиксации был получен четкий преципитат моноклонального %МХ, но для обнаружения μ-тяжелой цепи выбранное разведение могло оказаться избыточным. В результате произошло частичное растворение преципитата, и μ-тяжелую цепь выявить не удалось. При повторной иммунофиксации было использовано меньшее разведение исследуемой сыворотки, и μ-тяжелая цепь была обнаружена. Ретроспективно при исследовании исходного образца сыворотки с минимальным разведением и без разведения этот белок удалось найти. Вероятно, вследствие структурного дефекта μ-тяжелая цепь образует недостаточно прочное соединение с антисывороткой.
Морфологическая и иммуногистохимическая диагностика по трепанобиоптату костного мозга является важным звеном в процессе установления диагноза. При БТЦ-μ инфильтрат в костном мозге подобен таковому при хроническом лимфоцитарном лейкозе. Сочетание лимфоплазмоцитарной инфильтрации костного мозга и экстранодального поражения наталкивает на размышления о «размытости» диагностических границ. С одной стороны, видна лимфоплазмоцитарная лимфома IgM+/IgG+ с рестрикцией одной из легких цепей иммуноглобулина и минимальной секрецией аналогичного парапротеина. С другой — БТЦ-μ или БТЦ-γ, при которых может быть не нарушена продукция легких цепей с рестрикцией одной из них, выявляемой иммуногистохимически. Необходимость иммуногистохимического этапа диагностики, проведение электрофореза необходимо дополнять в таких случаях методом иммунофиксации. Наличие мутации гена MYD88+, характерной морфологической картины в костном мозге с выявлением секреции PIgM λ, определяют обоснованность предположения о многоэтапном формировании патологического процесса с возникновением нескольких мутаций в рамках молекулярного патогенеза заболевания. Например, одной из альтернативных точек зрения в данном случае может быть диагностика лимфоплазмоцитарной лимфомы, PIgMλ с мутацией в гене MYD88, возникновением генетической аномалии μ-тяжелой цепи иммуноглобулина и возникновением компонента моноклональной гаммапатии с невысоким уровнем μ-парапротеина.
Тяжесть клинической картины больного в описанном наблюдении не соответствовала небольшому объему опухоли. Прогрессирующая полинейропатия, ортостатическая гипотензия, рестриктивная кардиомиопатия, проявлявшаяся тяжелой сердечной недостаточностью, в совокупности с лимфоплазмоцитарной инфильтрацией костного мозга и секрецией легкой цепи λ указывали на системное амилоидное поражение, вероятно AL-амилоидоз. Сочетание БТЦ-μ c системным амилоидозом явление редкое. Описано всего три подобных случая [40].
AL-амилоидоз составляет 70% от всех случаев системного амилоидоза [41]. Морфологическим субстратом заболевания являются моноклональные легкие цепи, преимущественно λ-типа, откладывающиеся в виде депозитов в органах и тканях. Различают локальные формы с поражением сердца, почек, легких или мочевого пузыря и системный вариант заболевания. Медиана выживаемости варьирует от 4 месяцев у больных с хронической сердечной недостаточностью и до 50 месяцев у больных с периферической полинейропатией в дебюте заболевания [42].
Общая выживаемость может быть от 6 месяцев до 10 лет, медиана выживаемости больных с амилоидозом сердца напрямую зависит от показателей NT-proBNP, тропонина и соотношения вовлеченных и не вовлеченных в процесс легких цепей [43].
Несмотря на развернутую клиническую картину системного амилоидоза, в настоящем наблюдении не удалось прижизненно установить диагноз. Стандартная диагностика амилоидоза заключается в гистологическом исследовании биоптатов слизистой оболочки желудочно-кишечного тракта, подкожно-жировой клетчатки. Наиболее информативным и доступным является гистологическое исследование биоптата подкожно-жировой клетчатки передней брюшной стенки [44]. Выбор обусловлен минимальной инвазивностью и низким риском осложнений. Информативность исследования достигает 70%. Для исключения амилоидоза были взяты биоптаты десны, подкожно-жировой клетчатки, костного мозга и желудка, не было выявлено морфологических или гистохимических признаков отложения парапротеина.
Диагностика амилоидоза на основании исследования трепанобиоптата костного мозга имеет невысокую специфичность, в том числе из-за небольшого размера получаемого материала. В настоящем наблюдении этим можно объяснить обнаружение амилоида в отдельных сосудах костного мозга большого спила позвонка, взятом при аутопсии. Самым достоверным методом диагностики является биопсия пораженного органа. Очевидным было поражение сердца, и, учитывая наличие и характер протеинурии, можно было предположить поражение почек. Пункционная биопсия почки была сопряжена с большим риском кровотечения. Возможные осложнения при манипуляции могли усугубить течение болезни, а доказательство амилоидного поражения не изменило бы тактики терапии.
Особенностью клинического случая является сочетание БТЦ-μ с амилоидозом и массивными неамилоидными белковыми депозитами. Подобные редкие наблюдения были описаны японскими исследователями [40, 45]. В настоящем наблюдении отмечено сочетание амилоидных отложений: AL-амилоидоза и ATTR-амилоидоза с неамилоидным субстратом, возможно, принадлежащим к моноклональному отложению μ-иммуноглобулина — MIDD (monoclonal immunoglobulin deposition disease). При MIDD чаще откладывается κ-цепь иммуноглобулина и могут откладываться только тяжелые цепи или в сочетании с легкими цепями. Описано амилоидное поражение почки у больного с парапротеинемией IgM λ, протеинурией Бенс-Джонса λ, при иммунофлуоресцентном исследовании и иммуноэлектронной микроскопии в амилоидных отложениях определялась только μ-тяжелая цепь [46—48]. Если белковые отложения в органах приводят к их поражению, необходимо их детальное исследование с использованием электронной микроскопии, иммунофлюоресцентной микроскопии, масс-спектрометрии. Минимальное поражение почек в сочетании с массивным органным поражением (сердце, печень, селезенка, щитовидная железа, легкие, простата, брюшина) амилоидными и неамилоидными массами также являются особенностью данного наблюдения. Возможно, объяснением этого служит преобладание ATTR-амилоидоза.
Чаще всего транстиретиновый амилоидоз поражает печень, желудочно-кишечный тракт, селезенку. Известны два подтипа транстиретинового амилоидоза: немутантный («дикий») и наследственный (мутантный). «Дикий» тип чаще развивается у пожилых людей, и протекает с поражением сердца и развитием рестриктивной кардиомиопатии [49]. Мутантный тип подразделяется на наследственную семейную амилоидную полинейропатию и транстиретиновую кардиомиопатию. Заболевание развивается в результате мутации гена, кодирующего синтез транспортного белка транстиретина (TTR). В настоящее время известно около 100 подобных мутаций. Самые известные из них Val11Ile и Val30Met. Первую зачастую обнаруживают у больных с кардиальным вариантом заболевания, а вторая ассоциирована с амилоидной полинейропатией [49]. В 2013 г. были опубликованы данные регистра THAOS, в который были включены 957 больных с транстиретиновым амилоидозом, диагностированным в клиниках Португалии, США и Швеции. Было показано, что наследственный тип больше характерен для молодых больных. Заболеваемость уменьшается прямо пропорционально с увеличением возраста. Медиана возраста больных с впервые диагностированным амилоидозом составила 39 лет. У 885 из 957 больных, включенных в исследование, были выявлены характерные мутации, но только 611 больных имели клинические проявления транстиретинового амилоидоза. Таким образом, выделено 2 подгруппы больных: с симптоматическим и бессимптомным течением заболевания [50].
Молекулярно-генетическое исследование является важнейшим в диагностике транстиретинового амилоидоза. Из-за негативных результатов биопсии на обнаружение амилоида это исследование не было выполнено. В итоге установить конкретный тип заболевания было трудно.
В результате проведенных прижизненных исследований с расширенным спектром лабораторной диагностики и исследования аутопсийного материала у больного была установлена БТЦ-μ с морфологическим субстратом лимфоплазмоцитарной лимфо- мы в костном мозге, секрецией PIgM, монотипией λ-цепи, наличием экстранодального поражения паранефральной клетчатки, протекавшая с системным амилоидозом смешанного типа (ATTR, AL) и отложением неамилоидного белкового субстрата (возможно, MIDD) с поражением сердца, печени, селезенки, щитовидной железы, легких, предстательной железы, почек, брюшины. Непосредственной причиной смерти явилась сердечная недостаточность вследствие рестриктивной кардиомиопатии, обусловленная массивным отложением амилоидных и неамилоидных белковых масс в миокарде, стенках кровеносных сосудов.
Таким образом, диагноз БТЦ является трудным. Современные методы лабораторной и инструментальной диагностики не всегда позволяют быстро установить его. Основным критерием диагноза является наличие тяжелой цепи иммуноглобулина в сыворотке крови. Сложности при иммунохимическом исследовании могут быть связаны с низкой концентрацией тяжелой цепи в сыворотке. В настоящем наблюдении, несмотря на то что все симптомы указывали на системный амилоидоз, с помощью стандартных диагностических методов (биопсия слизистой оболочки желудочно-кишечного тракта, подкожно-жировой клетчатки, окраска трепанобиоптата костного мозга) был получен отрицательный результат. В редких случаях можно рассматривать вопрос о биопсии пораженного органа.
1. Bianchi G., Anderson K. C., Harris N.L., Sohani A.R. The heavy chain diseases: clinical and pathologic features. Oncology. 2014; 28(1): 45–53.
2. Андреева Н.Е., Балакирева Т.В. Болезни тяжелых цепей. Клиническая онкогематология: руководство для врачей. Под ред. М.А. Волковой. 2-е изд. М.: Медицина, 2007: 885–90.
3. Courtois L. Sujobert P. Morphologic features of μ-heavy-chain disease. Blood. 2017; 130(4): 558. DOI: 10.1182/blood-2017-04-781344.
4. Burmester G.-D., Pezzutto A. B-lymphocyte development and differentiation. In: Color atlas of immunology. Stuttgar; New York: Thieme. 2003: 32–46.
5. Witzig T.E., Wahner-Roedler D.L. Heavy chain disease. Curr Treat Opt Oncol. 2002; 3(3): 247–54. DOI: 10.1007/s11864-002-0014-3.
6. Ria R., Dammacco F., Vacca A. Heavy-chain diseases and myeloma-associated Fanconi syndrome: an update. Mediterr J Hematol Infect Dis. 2018; 10(1): e2018011. DOI: 10.4084/MJHID.2018.011.
7. Seligmann, M., Danon, F., Hurez, D., et al. Alpha-chain disease: a new immunoglobulin abnormality. Science. 1968; 162(3860): 1396–7. DOI: 10.1126/science.162.3860.1396.
8. Lecuit M., Abachin E., Martin A., et al. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N Engl J Med. 2004; 350(3): 239– 48. DOI: 10.1056/NEJMoa031887.
9. Fine K.D., Stone M.J. α-Heavy chain disease, Mediterranean lymphoma, and immunoproliferative small intestinal disease: a review of clinicopathological features, pathogenesis, and differential diagnosis Am J Gastroenterol. 1999; 94(5): 1139–52. DOI: 10.1111/j.1572-0241.1999.01057.x.
10. Kurimoto, M., Sonoki, T., Nakamura, Y., et al. Severe enteropathy caused by α-heavy chain disease lacking detectable M-proteins. Intern Med. 2014; 53(6): 581–5. DOI: 10.2169/internalmedicine.53.1625.
11. Franklin E.C., Lowenstein, J., Bigelow, B., et al. Heavy chain disease — a new disorder of serum γ-globulins: report of the fi rst case. Am J Med. 1964; 37(3): 332–50. DOI: 10.1016/0002-9343(64)90191-3.
12. Wahner-Roedler D.L., Kyle R.A. Heavy chain diseases. Best Pract Res Clin Haematol. 2005; 18: 729–46. DOI: 10.1016/j.beha.2005.01.029.
13. Fermand J.P., Brouet, J.C., Danon, F., et al. Gamma heavy chain “disease”: heterogeneity of the clinicopathologic features. Report of 16 cases and review of the literature. Medicine. 1989; 68(6): 321–35. DOI: 10.1097/00005792-198911000-00001.
14. Wahner-Roedler D.L. Witzig, T.E., Loehrer, L.L., et al. γ-Heavy chain disease: review of 23 cases. Medicine. 2003; 82(4): 236–50. DOI: 10.1097/01.md.0000085058.63483.7f.
15. San-José P., Aguadero V., Perea G., et al. Gamma heavy-chain disease accompanied with follicular lymphoma: a case report. Biochem Med. 2018; 28(1): 010802. DOI: 10.11613/BM.2018.010802.
16. Ramasamy, I., Rudzki, Z. Two Cases of γ-Heavy Chain Disease and a Review of the Literature Case Rep Hematol. 2018; 4832619. DOI: 10.1155/2018/4832619.
17. Cook J.R., Harris N.L., Isaacson P.G., et al. Heavy chain diseases. In: S.H. Swerdlow, E Campo, N.L., Harris, et al., eds. WHO Classifi cation of Tumours of Haematopoietic and Lymphoid Tissues. Revised 4th Edition. Lyon: IARC. Press; 2017: 238–9.
18. Van Keer J., Meijers B., Delforge M., et al. Two cases of heavy chain MGUS. Case reports in oncological medicine. 2016; 8749153. DOI: 10.1155/2016/8749153.
19. Forte F.A., Prelli F., Yount W.J., et al. Heavy chain disease of the μ (γM) type: Report of the fi rst case. Blood. 1970; 36(2): 137–44. DOI: 10.1182/blood. v36.2.137.137
20. Ballard H.S., Hamilton L.M., Marcus A.J., Illes C.H. A new variant of heavychain disease (μ-chain disease). N Engl J Med. 1970; 282(19): 1060–2. DOI: 10.1056/nejm197005072821902.
21. Haas I.G., Wabl M. Immunoglobulin heavy chain binding protein. Nature. 1983; 306: 387–9. DOI: 10.1038/306387a0.
22. Alexander A., Anicito I., Buxbaum J. Gamma heavy chain disease in man: genomic sequence reveals two noncontiguous deletions in a single gene. J Clin Invest. 1988; 82: 1244–52. DOI: 10.1172/JCI113722.
23. Leach I.H., Jenkins J.S., Murray-Leslie C.F., Powell R.J. μ-Heavy chain and monoclonal IgG K paraproteinaemia in systemic lupus erythematosus. Rheumatology. 1987; 26(6): 460–2. DOI: 10.1093/rheumatology/26.6.460.
24. Gordon J., Hamblin T. J., Smith J. L., et al. A human b-cell lymphoma synthesizing and expressing surface mu-chain in the absence of detectable light chain. Blood. 1981; 58(3): 552–6. DOI: 10.1182/blood.V58.3.552.bloodjournal583552.
25. Bonhomme J., Seligmann M., Mihaesco C., et al. Mu-chain disease in an African patient. Blood. 1974; 43(4): 485–92. DOI: 10.1182/blood.V43.4.485.485.
26. Witzens M., Egerer G., Stahl D., et al. A case of μ heavy-chain disease associated with hyperglobulinemia, anemia, and a positive Coombs test. Ann Hematol. 1998; 77(5): 231–4. DOI: 10.1007/s002770050448.
27. Wahner-Roedler D. L., Kyle R. A. μ-heavy chain disease: Presentation as a benign monoclonal gammopathy. Am J Hematol. 1992; 40(1): 56–60. DOI: 10.1002/ajh.2830400112.
28. Dammacco F., Bonomo L., Franklin E.C. A new case of mu heavy chain disease: clinical and immunochemical studies. Blood. 1974; 43(5): 713–9.
29. Yanai M., Maeda A., Watanabe N., et al. Successful treatment of μ-heavy chain disease with fludarabine monophosphate: a case report. Int J Hematol. 2004; 79(2): 174–7. DOI: 10.1532/IJH97.03053.
30. Guglielmo P., Granata P., Raimondo F.D., et al. ‘μ’ Heavy Chain Type ‘NonExcretory’ Myeloma. Scand J Haematol. 1982; 29(1): 36–40. DOI: 10.1111/j.1600-0609.1982.tb00559.x.
31. Leglise M.C., Briere J., Abgrall J.F., et al. Non-secretory myeloma of heavy mu-chain type. Nouv Rev Fr Hematol. 1983; 25(2): 103–6.
32. Maeda A., Mori M., Torii S., et al. Multiple Extranodal Tumors in μ-Heavy Chain Disease. Int J Hematol. 2006; 84(3): 286. DOI: 10.1532/IJH97.06124.
33. Vilpo J. A., Irjala K., Viljanen M. K., et al. δ-Heavy chain disease: A study of a case. Clin Immunol Immunopathol. 1980; 17(4): 584–94. DOI: 10.1016/0090-1229(80)90154-3.
34. Пирадов М.А., Супонева Н.А., Гинзберг М.А. и др. POEMS-синдром: обзор литературы и описание клинических наблюдений. Журнал неврологии и психиатрии им. СС Корсакова. 2014; 114(4): 4–10.
35. Балакирева Т.В., Андреева Н.Е. Макроглобулинемия Вальденстрема. Клиническая онкогематология. Фундаментальные исследования и клиническая практика. 2009; 2(2): 121–36.
36. Gertz M.A., Kyle R.A. Amyloidosis with IgM monoclonal gammopathies. Seminars in oncology 2003; 30(2): 325–8. DOI: 10.1053/sonc.2003.50060.
37. Ghobrial I.M. Are you sure this is Waldenström macroglobulinemia? Hematology 2010, the American Society of Hematology Education Program Book. 2012(1): 586–94. DOI: 10.1182/asheducation-2012.1.586.
38. Gertz M.A. Waldenström macroglobulinemia: 2017 update on diagnosis, risk stratifi cation, and management. Am J. Hematol. 2017; 92(2): 209–17. DOI: 10.1002/ajh.25292.
39. Corcos D., Osborn M.J., Matheson L.S. B-cell receptors and heavy chain diseases: guilty by association? Blood. 2011; 117(26): 6991–8. DOI: 10.1182/blood-2011-02-336164.
40. Kinoshita K., Yamagata T., Nozaki Y., et al. μ-Heavy Chain Disease Associated with Systemic Amyloidosis. Hematology. 2004; 9(2): 135–7. DOI: 10.1080/10245330410001671561.
41. Palladini G., Merlini G. What is new in diagnosis and management of light chain amyloidosis? Blood. 2016; 128(2): 159–68. DOI: 10.1182/blood-2016-01-629790.
42. Milani P., Merlini G., Palladini G. Light chain amyloidosis. Mediterr J Hematol Infect Dis. 2018; 10(1): e2018022. DOI: 10.4084/MJHID.2018.022.
43. Kumar S., Dispenzieri A., Lacy M. Q., et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin. Oncol. 2012; 30(9): 989. DOI: 10.1200/JCO.2011.38.5724.
44. De Larrea C.F., Verga L., Morbini P., et al. A practical approach to the diagnosis of systemic amyloidoses. Blood. 2015; 125(14): 2239–44. DOI: 10.1182/blood-2014-11-609883.
45. Manabe S., Iwasaki C., Hatano M., et al. AL amyloidosis with non-amyloid forming monoclonal immunoglobulin deposition; a case mimicking AHL amyloidosis. BMC nephrol. 2018; 19(1): 337. DOI: 10.1186/s12882-018-1050-y.
46. Mai H.L., Sheikh-Hamad D., Herrera G.A., et al. Immunoglobulin heavy chain can be amyloidogenic: morphologic characterization including immunoelectron microscopy. Am J Surg Pathol. 2003; 27(4): 541–5. DOI: 10.1097/00000478-200304000-00016.
47. Miyazaki D. Yazaki M., Gono T., et al. AH amyloidosis associated with an immunoglobulin heavy chain variable region (VH1) fragment: a case report. Amyloid. 2008; 15(2): 125–8. DOI: 10.1080/13506120802006229.
48. Yazaki M., Fushimi T., Tokuda T., et al. A patient with severe renal amyloidosis associated with an immunoglobulin γ-heavy chain fragment. Am J Kid Dis. 2004; 43(5): e22-1. DOI: 10.1053/j.ajkd.2003.12.056.
49. Дюдина И.А. Транстиретиновый амилоидоз: современное состояние проблемы. Серцева недостатність та коморбідні стани. 2017; 1: 63–8.
50. Coelho T., Maurer M. S., Suhr O. B. THAOS–The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013; 29(1): 63–76. DOI: 10.1185/03007995.2012.754348.
Охота Валерия Константиновна, врач-гематолог консультативного гематологического отделения с дневным стационаром по проведению интенсивной высокодозной химиотерапии
тел.: +7 (925) 128-79-76
Охота В.К., Рыжко В.В., Ковригина А.М., Шуплецова И.А., Соболева Н.П., Грибанова Е.О. Болезнь тяжелых цепей-μ в сочетании с системным амилоидозом и неамилоидными депозитами. Трудности диагностики и терапии. Гематология и трансфузиология. 2020;65(2):190-207. https://doi.org/10.35754/0234-5730-2020-65-2-190-207
Okhota V.K., Ryzhko V.V., Kovrigina A.M., Shupletsova I.A., Soboleva N.P., Gribanova E.O. μ-Heavy chain disease associated with systemic amyloidosis and non-amyloid deposits. Diffi culties in diagnosis and therapy. Russian journal of hematology and transfusiology. 2020;65(2):190-207. (In Russ.) https://doi.org/10.35754/0234-5730-2020-65-2-190-207
![]()
125167, Москва, Новый Зыковский проезд, 4
ФГБУ «НМИЦ гематологии» Минздрава России
тел.: 8-926-816-3887
e-mail: o.levchenko@htjournal.ru






































