

Содержание
Перейти к:
 А. И. Кашлакова,
Е. Н. Паровичникова,
Б. В. Бидерман,
Ю. В. Сидорова,
Ю. А. Чабаева,
В. В. Троицкая,
И. А. Лукьянова,
А. В. Кохно,
А. Н. Соколов,
А. Б. Судариков,
Т. Н. Обухова,
В. Г. Савченко
А. И. Кашлакова,
Е. Н. Паровичникова,
Б. В. Бидерман,
Ю. В. Сидорова,
Ю. А. Чабаева,
В. В. Троицкая,
И. А. Лукьянова,
А. В. Кохно,
А. Н. Соколов,
А. Б. Судариков,
Т. Н. Обухова,
В. Г. Савченко https://doi.org/10.35754/0234-5730-2020-65-4-444-459
Перейти к:
Введение. У больных острыми миелоидными лейкозами (ОМЛ) выявляются множественные драйверные мутации. Прогностическая значимость выявляемых мутаций в настоящее время до конца не изучена.
Цель: оценить частоту мутаций различных генов и их влияние на прогноз заболевания у взрослых больных ОМЛ.
Материалы и методы. В исследование включены 90 взрослых больных с впервые установленным диагнозом ОМЛ, из них 76 больных моложе 60 лет и 14 больных в возрасте от 60 лет и старше. В качестве индукции ремиссии у больных моложе 60 лет использовали программу химиотерапии (ХТ) «7+3», у больных старшей возрастной группы различные программы низкодозной ХТ с гипометилирующими агентами. Определение молекулярно-генетического статуса больных проводилось с использованием метода секвенирования нового поколения; авторская панель содержала гены ASXL1, BCOR, DNMT3, FLT3, IDH1, IDH2, PIGA, RUNX1, SETBP1, SF3B1, SRSF2, TET2, TP53 и U2AF2.
Результаты. Нуклеотидные замены были обнаружены в генах DNMT3, TET2, TP53, SETBP1, BCOR, RUNX1, IDH2, IDH1, FLT3, U2AF2, SF3B1 у 57,8 % больных (n = 52), при этом у 17,8 % больных (n = 16) были выявлены сочетанные мутации в двух или трех генах. Эффективность лечения и долгосрочные результаты терапии оценивали в зависимости от возраста, группы риска по ELN 2017 и наличия мутаций в генах TP53, RUNX1, IDH1, IDH2 и DNMT3. При оценке долгосрочных результатов терапии были выявлены достоверные различия в общей выживаемости (ОВ) в зависимости от наличия мутаций генов TP53 и RUNX1: у больных с мутациями TP53 ОВ составила 30 %, у больных без мутаций 53,4 % (p = 0,0037). Схожие результаты были получены для мутаций гена RUNX1 — у больных с мутациями ОВ составила 20 %, в то время как у больных без — 54 % (p = 0,0466).
Заключение. При диагностике ОМЛ определяют мутации в генах FLT3-ITD, NPM1 и CEBPA, однако генетическая гетерогенность группы больных ОМЛ диктует необходимость детального изучения их молекулярно-генетического статуса, что необходимо для уточнения прогноза заболевания и выбора оптимальной тактики терапии.
Кашлакова А.И., Паровичникова Е.Н., Бидерман Б.В., Сидорова Ю.В., Чабаева Ю.А., Троицкая В.В., Лукьянова И.А., Кохно А.В., Соколов А.Н., Судариков А.Б., Обухова Т.Н., Савченко В.Г. Определение молекулярно-генетического профиля у взрослых больных острыми миелоидными лейкозами методом секвенирования нового поколения. Гематология и трансфузиология. 2020;65(4):444-459. https://doi.org/10.35754/0234-5730-2020-65-4-444-459
Kashlakova A.I., Parovichnikova E.N., Biderman B.V., Sidorova Y.V., Chabaeva Y.A., Troitskaya V.V., Lukianova I.A., Kokhno A.V., Sokolov A.N., Sudarikov A.B., Obukhova T.N., Savchenko V.G. Next-generation sequencing-based molecular genetic profiling in adults with acute myeloid leukaemia. Russian journal of hematology and transfusiology. 2020;65(4):444-459. (In Russ.) https://doi.org/10.35754/0234-5730-2020-65-4-444-459
В последние годы в клинической практике у больных острыми миелоидными лейкозами (ОМЛ) выполняются молекулярно-генетические исследования. В определении групп риска у больных ОМЛ основополагающими методами являются стандартное цитогенетическое исследование и метод флуоресцентной гибридизации in situ (Fluorescence In Situ Hybridization, FISH). Изучено влияние на прогноз заболевания таких устойчивых хромосомных аномалий, как t(15;17)(q22;q12); PML-RARA, t(8;21)(q22;q22)/RUNX1-RUNX1T1 и inv(16)(p13q22)/t(16;16)(p13;q22)/CBFB-MYH11, выделенных в группу благоприятного прогноза, а также -5/(del)5q, -7, -17/abn(17p), inv(3)(q21.3q26.2)/t(3;3)(q21.3;q26.2) и комплексного кариотипа, являющихся, напротив, факторами неблагоприятного прогноза [1][2][3][4]. Однако у 50 % больных при цитогенетическом исследовании определяется нормальный кариотип, и в этом случае они стратифицируются в группу промежуточного риска [1]. Отсутствие у таких больных каких-либо цитогенетических аномалий затрудняет прогнозирование течения заболевания и не позволяет выбрать оптимальную тактику лечения. С появлением новых методов молекулярно-генетического анализа стало возможным рассматривать ОМЛ не только как цитогенетически гетерогенную группу заболеваний, но и как сложную, динамическую систему, характеризующуюся множественными соматическими драйверными мутациями и постоянной эволюцией сосуществующих опухолевых клонов [5]. За последние 20 лет было идентифицировано множество мутаций, ассоциированных с миелоидными новообразованиями, в частности с ОМЛ, и предпринято немало попыток создания классификации ОМЛ, учитывающей как биологическую природу опухоли, так и влияние выявляемых генетических поломок на дальнейший прогноз заболевания. В классификацию ВОЗ 2016 г. была добавлена категория «ОМЛ с генетическими мутациями» (AML with gene mutations), включающая такие варианты заболевания, как ОМЛ с мутацией NPM1, ОМЛ с биаллельной мутацией CEBPA и ОМЛ с мутацией RUNX1 (предварительная категория) [6]. Включение данных мутаций в классификацию ВОЗ обусловлено их частым выявлением у больных ОМЛ (20–45 % для NPM1, 5–10 % для CEBPA, 5–15 % для RUNX1), доказанной прогностической значимостью (мутации NPM1 и биаллельная мутация CEBPA являются факторами благоприятного прогноза, в то время как мутации RUNX1 — неблагоприятного), а также их специфической ролью в патогенезе опухоли [7][8][9][10][11][12][13][14][15][16]. Таким образом, классификация ВОЗ учитывает как биологические, так и клинические особенности ОМЛ с вышеуказанными мутациями.
В клинической практике наибольшим значением обладает молекулярно-генетическая стратификация ОМЛ по группам риска, предложенная в клинических рекомендациях European Leukemia Net (ELN) в 2017 г. В данной классификации учитываются мутации в генах NPM1, CEBPA (биаллельная мутация), а также в генах FLT3-ITD, RUNX1, ASXL1 и TP53 [5]. Мутации в генах RUNX1, ASXL1 и TP53, согласно данным многочисленных исследований, являются факторами неблагоприятного прогноза [17–25], мутации в генах NPM1 и CEBPA-biallelic ассоциированы с благоприятным прогнозом. Что касается мутаций в гене FLT3-ITD, значение имеет не только их наличие, но и уровень аллельного соотношения мутантного типа к «дикому» типу, а также отсутствие или наличие при этом мутации в гене NPM1. Мутации гена FLT3-ITD являются одними из наиболее часто выявляемых мутаций при ОМЛ, обнаруживаются у 25–30 % больных и ассоциированы с высоким риском развития рецидива [26][27][28][29][30][31][32]. Выявление мутаций FLT3-ITD в настоящее время является одним из важных прогностических маркеров при ОМЛ, так как позволяет не только прогнозировать течение заболевания, но и выбирать терапевтическую тактику с учетом появления нескольких таргетных препаратов.
Однако принятые классификации разработаны с учетом весьма ограниченного количества мутаций. Эти генетические аномалии являются наиболее изученными при ОМЛ и обладают вполне однозначной прогностической значимостью. Тем не менее существуют исследования, свидетельствующие о важности и других мутаций. Поиск новых терапевтических возможностей и прогностических критериев делает актуальным в настоящее время применение полногеномного секвенирования у больных ОМЛ.
В программе TCGA (The Cancer Genome Atlas) исследовательской группой ОМЛ были проведены полногеномное (n = 50) и полноэкзомное (n = 150) cеквенирования 200 случаев de novo ОМЛ и выявлены мутации суммарно в 260 генах; из них 23 гена были мутированы повсеместно, и в 237 генах мутации выявлялись лишь в двух или более случаях. Обнаруженные мутации были разбиты на 9 функциональных классов: мутации в генах, кодирующих нуклеофосмин (NPM1), мутации в генах опухолевой супрессии (TP53), мутации в генах эпигенетической регуляции, или генах, связанных с метилированием ДНК (DNMT3A, TET2, IDH1, IDH2), в генах сигнальных путей (FLT3, SETBP1), в генах, кодирующих факторы транскрипции (RUNX1, CEBPA), в генах-модификаторах хроматина (ASXL1, BCOR), в генах-факторах сплайсинга (SRSF2, SF3B1, U2AF1, U2AF2) и в генах двух других классов (гены, кодирующие белки когезина, и химерные гены) [7]. Роль перечисленных мутаций в лейкемогенезе и их влияние на прогноз заболевания в настоящий момент изучаются. Несмотря на неоднозначность и противоречивость имеющихся данных, некоторые исследователи предлагают выделить дополнительные категории ОМЛ, помимо описанных в классификации ВОЗ 2016 г. E. Papaemmanuil и соавт. [33], проанализировав клинические, цитогенетические и молекулярно-генетические данные 1540 больных ОМЛ, разделили ОМЛ на 11 непересекающихся классов, каждый со своими клиническими характеристиками и прогнозом. Среди новых предложенных категорий — ОМЛ с мутациями в генах-модификаторах хроматина и генах-факторах сплайсинга, ОМЛ с мутациями гена TP53 и/или анеуплоидией, ОМЛ с мутациями гена IDH2 в кодоне R172. Таким образом, несмотря на прогресс в изучении молекулярной биологии ОМЛ, неясны все патогенетические механизмы и закономерности течения данного заболевания.
Цель настоящего исследования — оценить частоту мутаций различных генов и их влияние на прогноз заболевания у взрослых больных ОМЛ.
В биобанке лаборатории молекулярной гематологии ФГБУ «НМИЦ гематологии» Минздрава России накоплен первичный материал (аспират костного мозга, взятый в дебюте заболевания) от взрослых больных с впервые установленным диагнозом ОМЛ, поступивших в Центр в период с 2012 г. по 2019 г. Для решения поставленной задачи были отобраны образцы ДНК от 100 больных, выделенные в период с 2016 г. по 2018 г. Впоследствии 10 больных были исключены из дальнейшего анализа в связи с недостаточным количеством материала для проведения исследования. Таким образом, в исследование были включены 90 больных ОМЛ.
Все больные были разделены на две возрастные группы: моложе 60 лет и от 60 лет и старше. Группу больных моложе 60 лет составили 76 человек (соотношение мужчин и женщин — 23 : 53), группу больных от 60 лет и старше — 14 человек (соотношение мужчин и женщин — 8 : 6). Медиана наблюдения составила 20,9 месяца (0,07–78,3 месяца).
Диагноз ОМЛ у всех больных был установлен на основании исследования общего анализа крови, а также цитологического, цитохимического, иммунофенотипического, цитогенетического и молекулярно-генетического исследований аспиратов костного мозга. Клинико-лабораторная характеристика больных ОМЛ в зависимости от возраста представлена в таблице 1.
Таблица 1. Клинико-лабораторная характеристика больных в зависимости от возраста
Table 1. Clinical and laboratory profile of age cohorts
Возраст, лет Age, years | < 60 (n = 76) | ≥ 60 (n = 14) |
Пол Gender, n (%) • Мужчины / Male • Женщины / Female | 23 (30,3) 53 (69,7) | 8 (57,1) 6 (42,9) |
Группы риска по ELN 2017, n (%) ELN-2017 risk cohorts, n (%) | ||
• Благоприятный • Favourable | 18 (23,7) | 1 (7,1) |
• Промежуточный • Intermediate | 37 (48,7) | 6 (42,9) |
• Неблагоприятный • Unfavourable | 21 (27,6) | 7 (50) |
Цитогенетические аберрации и данные молекулярного исследования, n (%) Cytogenetic aberrations and molecular evidence, n (%) | ||
| ||
• t(8;21)(q22;q22.1); RUNX1-RUNX1T1 | 6 (7,9) | – |
• inv(16)(p13.1q22); CBFB-MYH11 | 3 (4) | – |
• Нормальный кариотип + мутация в гене NPM1 • Normal karyotype + NPM1 mutation | 9 (11,8) | 1 (7,1) |
| ||
• (9;11)(p21.3;q23.3); MLLT3-KMT2A | 1 (1,3) | – |
• Неклассифицируемые численные аномалии • Unclassified numerical abnormalities | 6 (7,9) | 2 (14,3) |
• Неклассифицируемые структурные аномалии • Unclassified structural abnormalities | 2 (2,6) | 1 (7,1) |
• Нормальный кариотип + мутации в генах NMP1 и FLT3-ITD • Normal karyotype + NPM1 or FLT3-ITD mutations | 5 (6,6) | 1 (7,1) |
• Нормальный кариотип без мутаций NPM1, FLT3-ITD • Normal karyotype, no NPM1 or FLT3-ITD mutations | 23 (30,2) | 2 (14,3) |
| ||
• –7/del(7q) | 5 (6,6) | – |
• Комплексный или моносомный кариотип • Complex or monosomal karyotype | 9 (11,8) | 5 (35,8) |
• Нормальный кариотип + мутация в гене FLT3-ITD • Normal karyotype + FLT3-ITD mutation | 4 (5,3) | – |
• Другое, в т. ч. сочетанные аномалии • Other, including combined abnormalities | 3 (4) | 2 (14,3) |
• Концентрация гемоглобина крови в дебюте, медиана (диапазон), г/л • Haemoglobin concentration at debut, median (range), g/L | 84 (50–142) | 85 (49–135) |
• Количество тромбоцитов крови в дебюте, медиана (диапазон), × 109/л • Platelet count at debut, median (range), × 109/L | 57 (3–417) | 56,5 (17–189) |
• Количество бластных клеток в костном мозге дебюте, медиана (диапазон), % • Blast cell count in bone marrow at debut, median (range), % | 65,8 (10,8–96,8) | 65,2 (23,2–92,8) |
В качестве индукции ремиссии использовали различные программы химиотерапии (ХТ). В группе больных моложе 60 лет у большинства больных применяли программу ХТ по схеме «7+3», предусматривающую непрерывную инфузию цитарабина в дозе 200 мг/м2/сут. в течение 7 дней и введение в течение 3 дней антрациклинового антибиотика (даунорубицина либо идарубицина). Впоследствии 48,7 % больных (n = 37) из данной возрастной группы была выполнена трансплантация аллогенных гемопоэтических стволовых клеток (алло-ТГСК). В старшей возрастной группе у большинства больных применяли курсы низкодозной химиотерапии с гипометилирующими агентами. Алло-ТГСК этим больным не выполняли. Характеристика применявшихся программ лечения представлена в таблице 2.
Таблица 2. Характеристика программ лечения в зависимости от возраста
Table 2. Therapy structure in age cohorts
Программы индукционной ХТ Induction chemotherapy | Больные < 60 лет Patients < 60 years (n = 76) | Больные ≥ 60 лет Patients ≥ 60 years (n = 14) |
Программы стандартной ХТ Standard chemotherapy | ||
«7+3» — n (%) | 61 (80,3) | 4 (28,6) |
«7+3» с предфазой Aza — n (%) “7+3” + pre-phase with Aza, n (%) | 8 (10,5) | – |
Курсы низкодозной ХТ Low-dose chemotherapy | ||
Aza-Ida-Ara-C, n (%) | 6 (7,9) | 4 (28,6) |
Dac-Ida-Ara-C, n (%) | 1 (1,3) | 1 (7,1) |
Aza + Венетоклакс, n (%) Aza + Venetoclax, n (%) | – | 3 (21,4) |
Без лечения, n (%) Intact, n (%) | – | 2 (14,3) |
Алло-ТГСК — n (%) Allo-HSCT, n (%) | 37 (48,7) | 0 |
• в первой ремиссии, n • first complete remission, n | 31 | |
• во второй ремиссии, n • second complete remission, n | 2 | – |
• вне ремиссии, n • no remission, n | 4 | |
Для проведения секвенирования нового поколения использовали технологию таргетного обогащения библиотек фрагментов ДНК HEAT-SEQ (Roche Diagnostics, Швейцария). Эта система основана на амплификации таргетных генов с использованием инвертированных молекулярных зондов для детекции различных вариантов исследуемых мишеней. Авторская панель Myelo_set_HEAT была подобрана на основании литературных данных и содержала гены ASXL1 (экзон 12), BCOR (экзоны 1–14), DNMT3 (экзоны 7–23), FLT3 (экзоны 13–16, 20), IDH1 (экзон 4), IDH2 (экзон 4), NPM1 (экзоны 10–11), PIGA (экзоны 1–5), RUNX1 (экзоны 4–9), SETBP1 (экзон 4), SF3B1 (экзоны 14–16), SRSF2 (экзон 1), TET2 (экзоны 3–11), TP53 (экзоны 4–10), U2AF2 (экзоны 2, 6) [34]. Анализ нуклеотидных последовательностей производили на полногеномном секвенаторе MySeq (Illumina, США) с использованием наборов MiSeq Reagents Kit v2 — 300 cycles (Illumina, США). Фильтрование данных, удаление служебных последовательностей, картирование прочтений, поиск вариантов и удаление повторов осуществляли при помощи утилит FastQC, SAMtools, trimmomatic, hsqutils и bwa. Полученные результаты анализировали с помощью свободного программного обеспечения wANNOVAR [35]. Информация о патогенности мутаций была получена из открытых баз данных COSMIC [36] и DB SNP.
Для определения мутаций генов NPM1, FLT3-ITD и CEBPA были использованы последовательности праймеров, описанные ранее [37][38]. Чтобы избежать аденилирования (присоединения лишнего аденина) при работе Taq-полимеразы и возникновения эффекта «двойных» пиков при капиллярном электрофорезе, на 5’ конце обратных праймеров была помещена нуклеотидная последовательность gtttctt, в соответствии с рекомендациями [39]. Для полимеразной цепной реакции (ПЦР) использовали реактивы ЗАО Синтол, Москва. 20 мкл реакционной смеси содержало: 100–200 нг ДНК, 5 пмоль прямого и обратного праймеров, dNTP (0,25 мМ), 10 × ПЦР-буфер, MgCl2 (2 мМ), DMSO 1,5 мкл, Taq-полимеразу (1,25 ед.). Условия ПЦР: 95° 7 мин, затем 30 циклов — 95° 45 сек, 60° 45 сек, 72° 60 сек, затем окончательная элонгация 72° 5 мин. ПЦР проводили на амплификаторе T100 (Bio-Rad Laboratories, США). Для капиллярного электрофореза и фрагментного анализа ПЦР-продуктов использовали автоматический анализатор нуклеиновых кислот ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, США). Для этого 2 мкл разведенного в 50–250 раз ПЦР-продукта смешивали с 10 мкл Hi-Di формамида (Applied Biosystems, США) и 0,04 мкл GeneScan 500-LIS Size Standard (Applied Biosystems, США). После денатурации при 95 °C в течение 3 минут и последующего охлаждения 10 мкл смеси наносили в лунку 96-луночной плашки и проводили капиллярный электрофорез высокого разрешения на полимере POP-4 (Applied Biosystems, США). Флуоресценция и распределение амплификатов по длине оценивались при помощи компьютерной программы GeneMapper v. 4.0 (Applied Biosystems, США).
Для исследования точечных соматических мутаций генов IDH1 (p.R132H, p.R132C/G/S) и IDH2 (p.R140Q; p.R172K) использовали аллель-специфичную ПЦР в реальном времени. 200 нг ДНК вносили в 25 мкл реакционной смеси (ЗАО Синтол, Москва), содержащей 10 пмоль прямого и обратного праймеров и 7,5 пмоль пробы. ПЦР проводили в дупликатах на приборе Step One Real-Time PCR (Applied Biosystems, США). Условия ПЦР: 95° 5 мин, затем 50 циклов — 95° 30 сек, 62° 20 сек, 72° 20 сек. В качестве отрицательных контролей использовали смесь ДНК здоровых доноров. В качестве положительных контролей — образцы с подтвержденными секвенированием по Сенгеру мутациями.
Статистический анализ. Для статистической обработки данных использовали стандартные методы описательного, частотного и событийного анализа. Расчеты проводили с помощью процедур пакета SAS 9.4. Анализ общей выживаемости и безрецидивной выживаемости проводили с использованием оценок Каплана — Мейера и логрангового критерия для сравнения групп. При проведении многофакторного анализа и оценки отношения рисков использовали модель пропорциональных рисков (модель Кокса). Для сравнения категориальных переменных использовали критерий χ2. При анализе общей выживаемости временной интервал отсчитывали от даты диагностики заболевания до даты смерти (событие)/даты последнего контакта (цензурирование). При анализе безрецидивной выживаемости временной интервал отсчитывали от даты ремиссии до даты смерти или рецидива (событие)/даты последнего контакта (цензурирование). Поскольку в работе оценивали эффективность всего протокола терапии, в котором алло-ТГСК является его частью, цензурирование на момент трансплантации не выполняли.
Все больные были распределены по группам риска, согласно клиническим рекомендациям ELN 2017 [5]: среди больных моложе 60 лет группа благоприятного риска составила 23,7 % (n = 18), группа промежуточного риска — 48,7 % (n = 37), группа неблагоприятного риска — 27,6 % (n = 21). Среди больных от 60 лет и старше к указанным группам риска было отнесено 7,1 % (n = 1), 42,9 % (n = 6) и 50 % (n = 7) больных соответственно.
В группе моложе 60 лет у 41 (54 %) больного определили нормальный кариотип. Из цитогенетических аберраций в группе благоприятного риска у 6 (7,9 %) больных была выявлена t(8;21)(q22;q22.1); RUNX1-RUNX1T1, у 3 (4 %) — inv(16)(p13.1q22); CBFB-MYH11. В группе промежуточного риска у одного больного была выявлена t(9;11)(p21.3;q23.3); MLLT3-KMT2A. У 8 (10,5 %) больных в этой прогностической группе выявлялись численные, либо структурные цитогенетические перестройки, не классифицируемые как благоприятные или неблагоприятные, среди которых наиболее часто выявлялась трисомия 8-й хромосомы (n = 4). В группе неблагоприятного риска у 9 больных (11,8 %) был выявлен комплексный или моносомный кариотип, у 7 (9,2 %) больных определили моносомию 7-й хромосомы или del(7q), при этом у 2 — в сочетании c del(5q) или перестройкой гена EVI.
В группе больных от 60 лет и старше у 4 (28,6 %) больных был определен нормальный кариотип. Цитогенетических перестроек в группе благоприятного риска выявлено не было. В группе промежуточного прогноза были выявлены трисомия 8-й хромосомы, а также другие численные и структурные перестройки, не классифицируемые как благоприятные или неблагоприятные. В группе неблагоприятного прогноза у 5 (35,8 %) больных был выявлен комплексный кариотип.
Распределение по группам риска проводили согласно данным цитогенетического и молекулярно-генетического исследований на этапе установки диагноза. При проведении молекулярного исследования устанавливали наличие мутаций в генах CEBPA, NPM1 и FLT3-ITD, оценка аллельного соотношения мутантного типа к «дикому» при наличии мутации в гене FLT3-ITD не проводилась. Таким образом, больные с нормальным кариотипом и изолированной мутацией в гене FLT3-ITD были определены в группу неблагоприятного прогноза; в случае наличия у больных с нормальным кариотипом сочетанных мутаций NPM1 и FLT3-ITD их определяли в группу промежуточного прогноза.
В ходе проведенного исследования нуклеотидные замены были обнаружены в генах DNMT3, TET2, TP53, SETBP1, BCOR, RUNX1, IDH2, IDH1, FLT3, U2AF2, SF3B1 у 52 (57,8 %) из 90 больных. При этом у 16 (17,8 %) больных выявлены сочетанные мутации в двух или трех генах. Спектр выявленных мутаций представлен на рисунке 1.
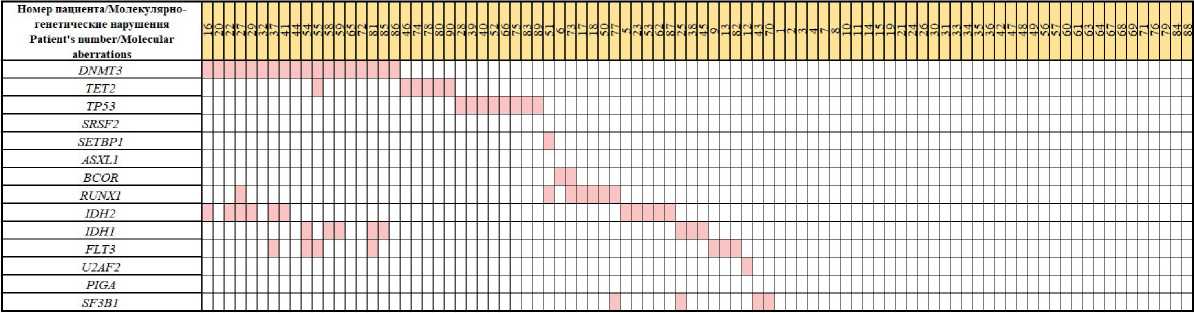
В гене TET2 выявлены 5 нонсенс-мутаций в 3-м, 10-м и 11-м экзонах (p.342X, p.454X, p.550X, p.R1465X и p.Q1942X) и 2 миссенс-мутации в 7-м (p.G1275R) и 9-м экзонах (p.R1359C). У 8 больных выявлены нуклеотидные замены в гене TP53. Из них 7 относятся к патогенным — в 5, 6, 7 и 8-м экзонах (p.Y126N, p.V157I, p.R175G, p.R196Q, p.Y220C, R248W, p.C275Y, p.R273H). В гене SETBP1 у одного больного выявлена миссенс-мутация p.V1200E неизвестного значения. Патогенная миссенс-мутация в 12-м экзоне гена BCOR (p.R1513X) выявлена у одного больного. Также выявлена нонсенс-мутация p.K1295X в 9-м экзоне данного гена, отсутствующая в известных базах данных. В 6-м экзоне гена RUNX1 у 2 больных обнаружены мутации в позиции R204 — миссенс-мутация p.R204Q и нонсенс-мутация p.R204X. Также выявлены 4 патогенные миссенс-мутации в 5-м экзоне — p.F158I, p.D160E, p.R162S, p.R166Q, и одна — в 6-м экзоне (p.S172G) с неизвестным клиническим значением. Патогенные мутации в генах IDH1 и IDH2 выявлены суммарно у 19 (21,1 %) из 90 больных. В 4-м экзоне гена IDH2 у 7 больных выявлена мутация p.R140Q, у 4 больных — p.R172K. В 4-м экзоне гена IDH1 у 5 больных обнаружена мутация p.R132C и еще у 2 — p.R132H. При выполнении аллель-специфичной ПЦР были подтверждены все выявленные мутации генов IDH1 и IDH2, а также выявлен еще один больной с мутацией гена IDH1 p.R132H. У 5 (5,6 %) больных выявлена мутация FLT3-TKD в 20-м экзоне (p.D835Y). Еще у 2 больных выявлена патогенная нуклеотидная замена в 14-м экзоне (p.V579E) и 16-м экзоне (p.K663R) гена FLT3. Мутации FLT3-ITD не исследовались в связи с особенностями методики. Патогенные мутации в 15-м экзоне гена SF3B1 выявлены у 4 больных и представлены заменой p.K700E. В 6-м экзоне гена U2AF2 обнаружена миссенс-мутация p.L175P неизвестного значения. В исследуемых экзонах генов SRSF2, ASXL1 и PIGA клинически значимых мутаций обнаружено не было. Характеристика выявленных мутаций в зависимости от возраста представлена в таблице 3.
Таблица 3. Молекулярно-генетический профиль больных в зависимости от возраста
Table 3. Molecular genetic profile of age cohorts
Ген, число больных (n) Gene, number of patients (n) | Нуклеотидные замены Nucleotide substitutions | Возраст, лет Age, years | |
< 60 (n = 76) | ≥ 60 (n = 14) | ||
DNMT3 (n = 18) | exon14:c.G1627T:p.G543C exon16:c.A1922G:p.D641G exon17:c.T1964C:p.I655T exon18:c.T2114C:p.I705T exon18:c.C2158T:p.R720C exon19:c.C2311T:p.R771X exon23:c.G2645A:p.R882H exon23:c.C2644T:p.R882C exon23:c.C2644A:p.R882S | n = 3 n = 1 n = 1 n = 1 – n = 1 n = 7 n = 2 n = 1 | – – – – n = 1 – – – n = 1 |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 16 (21) | 2 (14,3) | |
TET2 (n = 6) | exon3:c.A1360T:p.K454X exon3:c.G1024T:p.G342X exon3:c.C1648T:p.R550X exon7:c.G3823C:p.G1275R exon9:c.C4075T:p.R1359C exon10:c.C4393T:p.R1465X exon11:c.C5824T:p.Q1942X | – – n = 1 n = 1 n = 1 n = 1 n = 1 | n = 1 n = 1 – – – – – |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 4 (5,3) | 2 (14,3) | |
TP53 (n = 8) | exon5:c.G469A:p.V157I exon5:c.C523G:p.R175G exon6:c.G587A:p.R196Q | n = 1 n = 1 – – – – n = 1 – – | – – n = 1 n = 1 n = 1 n = 1 – n = 1 n = 1 |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 3 (4) | 5 (35,7) | |
SETBP1 (n = 1) | exon4:c.T3599A:p.V1200E | n = 1 | – |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 1 (1,3) | 0 | |
BCOR (n = 2) | exon9:c.A3883T:p.K1295X exon12:c.C4537T:p.R1513X | n = 1 n = 1 | – – |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 2 (2,6) | 0 | |
RUNX1 (n = 7) | exon5:c.C480A:p.D160E exon5:c.G486T:p.R162S exon5:c.T472A:p.F158I exon5:c.G497A:p.R166Q exon6:c.C610T:p.R204X exon6:c.G611A:p.R204Q exon6:c.A514G:p.S172G | n = 1 n = 1 n = 1 n = 1 n = 1 n = 1 n = 1 | – – – – – – – |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 7 (9,2) | 0 | |
IDH1 (n = 8) | exon4:c.C394T:p.R132C exon4:c.G395A:p.R132H | n = 4 n = 2 | n = 1 n = 1 |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 6 (7,9) | 2 (14,3) | |
IDH2 (n = 11) | exon4:c.G515A:p.R172K exon4:c.G419A:p.R140Q | n = 4 n = 7 | – – |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 11 (14,5) | 0 | |
FLT3 (n = 7) | exon14:c.T1736A:p.V579E | n = 1 – n = 5 | – n = 1 – |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 6 (7,9) | 1 (7,1) | |
U2AF2 (n = 1) | exon6:c.T524C:p.L175P | – | n = 1 |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 0 | 1 (7,1) | |
SF3B1 (n = 4) | exon15:c.A2098G:p.K700E | n = 4 | – |
Суммарно в группе, число больных (%) Cohort total, number of patients (%) | 4 (5,3) | 0 | |
Эффективность лечения оценивали по достижению полной ремиссии (ПР) после двух курсов индукционной ХТ. В группе больных моложе 60 лет ПР была достигнута у 64 (84,2 %) больных, у 6 (7,9 %) больных была констатирована первичная рефрактерность; ранняя летальность составила 6,6 % (n = 5). Данные о результатах лечения одной больной отсутствуют в связи с выпиской из стационара по собственному желанию. В группе больных от 60 лет и старше двое больных умерли до начала лечения. ПР была достигнута у 10 (83,3 %) больных, у 1 (8,3 %) констатирована первичная рефрактерность. Ранняя летальность в старшей возрастной группе составила 8,3 % (n = 1).
В группе благоприятного риска у всех 19 (100 %) больных была достигнута ПР. В группе промежуточного риска ПР была достигнута у 38 (88,4 %) больных, у одной больной констатирована первичная рефрактерность, данные об эффективности лечения у одной больной отсутствуют. Ранняя летальность составила 7 % (n = 3). В группе неблагоприятного риска у 17 (65,4 %) больных была достигнута ПР, у 6 (23,1 %) констатирована первичная рефрактерность; ранняя летальность составила 11,5 % (n = 3). Двое больных умерли до начала лечения.
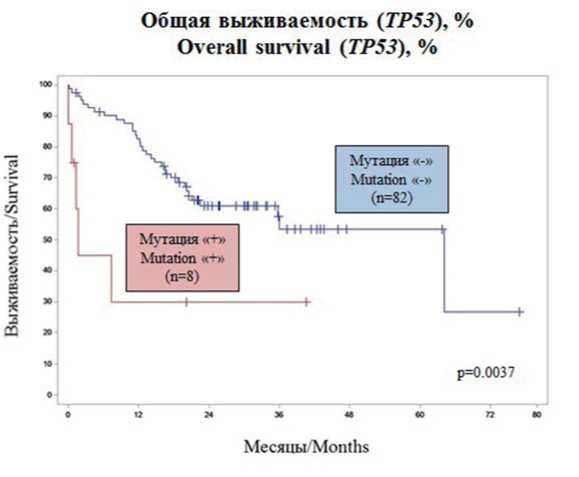
При анализе долгосрочных результатов терапии оценивали 3-летнюю общую выживаемость (ОВ) и безрецидивную выживаемость (БРВ) в разных возрастных группах (< 60 и ≥ 60 лет), в группах риска по ELN 2017 [5] и в зависимости от наличия у больных мутаций генов TP53, RUNX1, IDH1, IDH2 и DNMT3. ОВ в общей группе составила 51,6 %; в группе больных моложе 60 лет — 57,2 %, в группе больных от 60 лет и старше медиана наблюдения 36 месяцев достигнута не была, в связи с чем оценить 3-летнюю ОВ не удалось (p = 0,0006) (рис. 2). В зависимости от группы риска (благоприятный, промежуточный, неблагоприятный) ОВ составила 63,2, 69,5 и 18,6 % (p = 0,0001) (рис. 3). Статистически значимые различия в ОВ в зависимости от наличия тех или иных мутаций были выявлены для больных с мутацией генов TP53 и RUNX1: у больных с мутацией TP53 ОВ была ниже, чем у больных без нее (30 % против 53,4 %, p = 0,0037) (рис. 4). Наличие мутации в гене RUNX1 явилось фактором, неблагоприятно влияющим на ОВ: у больных с мутацией ОВ составила 20 %, в то время как у больных без нее — 54 % (p = 0,0466) (рис. 5).
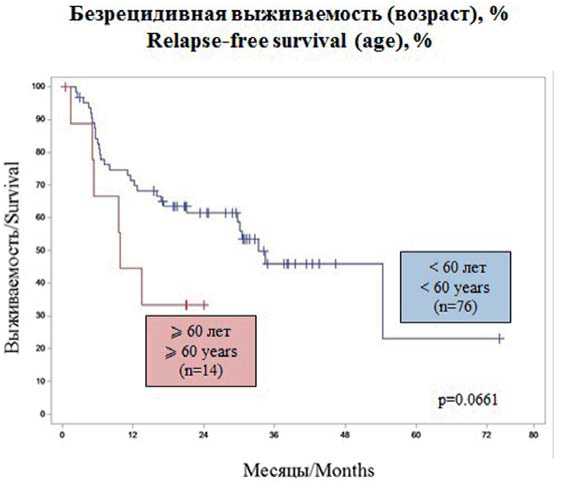
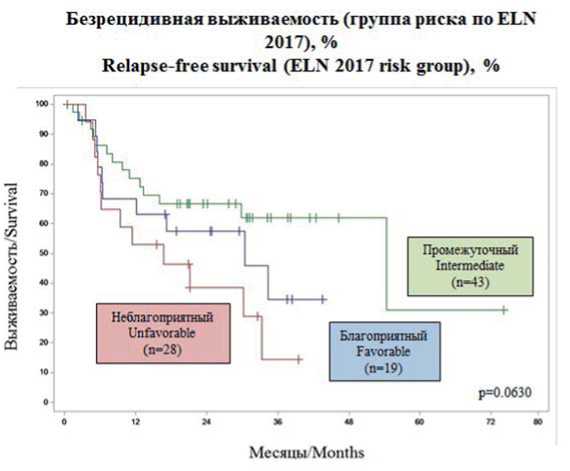
БРВ в общей группе составила 43,4 %, в группе больных моложе 60 лет — 46 % (p = 0,0661). В группе больных от 60 лет и старше не удалось оценить 3-летнюю ОВ в связи с меньшим сроком наблюдения за выжившими больными (рис. 6). В зависимости от группы риска БРВ составила 34,5, 62 и 14,5 % соответственно (p = 0,0630) (рис. 7). Достоверных различий в зависимости от наличия тех или иных мутаций при оценке БРВ выявлено не было.
При проведении многофакторного анализа влияния различных факторов на ОВ и БРВ с пошаговой селекцией в качестве факторов-кандидатов на вхождение в модель были включены мутация в гене TP53, мутация в гене RUNX1, а также группа риска по ELN 2017 в двух градациях (благоприятного/промежуточного либо неблагоприятного риска) и возраст (< 60 и ≥ 60 лет). Прогностическую значимость установили для следующих факторов: при оценке ОВ — группа риска по ELN 2017 и возраст, при оценке БРВ — только группа риска ELN 2017 [5].
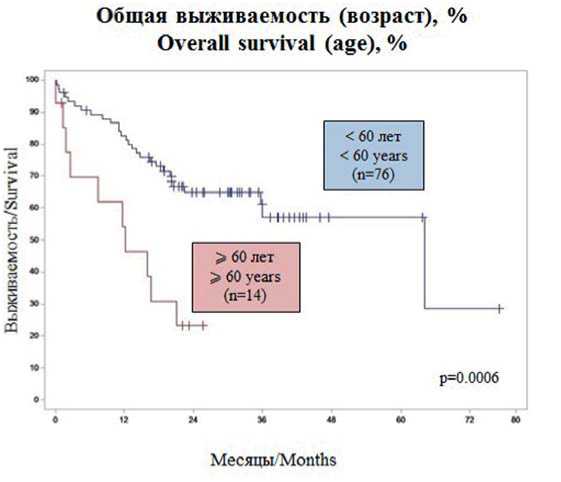
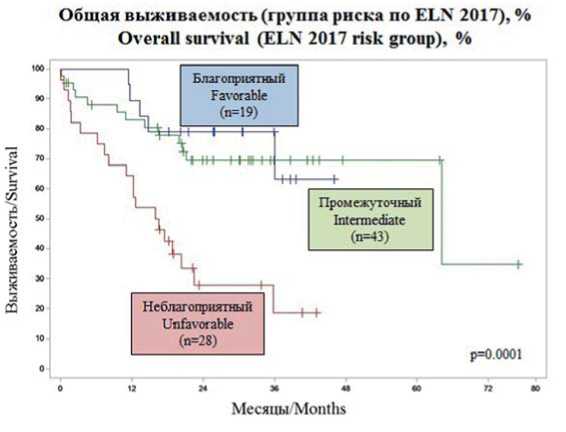

При проведении настоящего исследования у 88 % больных были выявлены молекулярные либо цитогенетические перестройки, что сопоставимо с данными крупных исследований, согласно которым более 90 % больных ОМЛ имеют, как минимум, одну драйверную мутацию [33][34]. Для решения поставленных задач была выбрана молекулярная панель из 14 генов, мутации в которых имеют прогностическую значимость при ОМЛ. В целом частота большей части выявленных мутаций оказалась сопоставима с частотой, описываемой в литературе. Мутации гена DNMT3 были выявлены у 20 % больных, мутации гена TP53 — у 9 % больных, RUNX1 — у 8 %, IDH1/IDH2 — у 21 % [33][40][41]. Некоторые из исследовавшихся мутаций, такие как ASXL1, SRSF2, SF3B1, U2AF2 и BCOR, характерны для вторичного ОМЛ и для ОМЛ, обусловленного предшествовавшей химиотерапией, более чем для de novo ОМЛ [42]. Возможно, этим объясняется отсутствие (ASXL1) либо низкая частота мутаций выбранных генов у больных в нашем исследовании.
Сравнение долгосрочных результатов терапии больных в зависимости от молекулярно-генетического профиля подтвердило неблагоприятное прогностическое значение мутаций таких генов, как TP53 и RUNX1. Показано, что в группе больных без мутаций TP53 ОВ была достоверно выше, чем в группе с мутациями (54 % против 30 %). При оценке эффективности терапии в группе больных без мутаций TP53 частота достижения ПР составила 85,4 %, первично-рефрактерное течение заболевания констатировано у 8,5 % больных, а ранняя летальность составила 4,9 %. Данные об эффективности лечения одной больной отсутствуют. В группе больных с мутацией гена TP53 лишь у 50 % больных была достигнута ПР. Смертность составила так же 50 %: двое больных умерли до начала лечения, двое — на этапах индукции ремиссии. Таким образом, мутации гена TP53 являются независимым прогностическим фактором, оказывающим неблагоприятное влияние как на эффективность терапии, так и на ее долгосрочные результаты. Подавляющее большинство больных (75 %) с выявленными мутациями TP53 были отнесены к группе неблагоприятного риска по ELN 2017 [5]: у 5 больных выявили комплексный кариотип, у 1 — моносомный. Наблюдалась ассоциация между наличием указанных мутаций и возрастом: 62,5 % больных с мутациями принадлежали к старшей возрастной группе (≥ 60 лет). Полученные результаты соответствуют данным литературы, согласно которым мутации гена TP53 ассоциированы с пожилым возрастом, сложными цитогенетическими поломками, в частности, с комплексным либо моносомным кариотипом, и плохим прогнозом [23]. Схожие результаты при оценке долгосрочных результатов терапии были получены для больных с мутациями гена RUNX1 (ОВ для больных с мутациями составила 20 % по сравнению с 54 % у больных без мутаций). Однозначность получаемых результатов в отношении мутаций TP53 и RUNX1 лишний раз доказывает необходимость их исследования еще на этапе установки диагноза ОМЛ. В то же время при оценке БРВ не было выявлено достоверных различий в зависимости от наличия тех или иных мутаций.
Особенность настоящего исследования заключается в расширении диапазона исследуемых мутаций при ОМЛ с использованием новейших молекулярно-генетических технологий, в том числе секвенирования нового поколения. Данный метод позволяет значительно повысить производительность и чувствительность исследования по сравнению с методом секвенирования по Сэнгеру. Проведение высокопроизводительного секвенирования с использованием соответствующих генетических панелей является критически важным для онкогематологических заболеваний, в частности ОМЛ, с учетом высокой частоты мутаций у данных больных и появлением все большего количества таргетных препаратов.
Таким образом, проанализирован материал от 90 больных с впервые установленным диагнозом ОМЛ с помощью секвенирования нового поколения. Данный метод позволяет в короткие сроки проанализировать массивный объем информации и получить развернутую характеристику молекулярно-генетического статуса для каждого больного. Полученные результаты свидетельствуют о необходимости дальнейшего изучения мутационного профиля ОМЛ с большим объемом выборки и проведением развернутого статистического анализа.
1. Grimwade D., Hills R.K., Moorman A.V. et al. Refinement of cytogenetic classification in AML Younger adult patients treated in UKMRC. Blood. 2010; 116(3): 354–66. DOI: 10.1182/blood-2009-11-254441.
2. Grimwade D., Walker H., Oliver F. et al. The importance of diagnostic cytogenetics on outcome in AML: Analysis of 1,612 patients entered into the MRC AML 10 trial. Blood. 1998; 92(7): 2322–33. DOI: 10.1182/blood.v92.7.2322.
3. Grimwade D., Walker H., Harrison G. et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): Analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001; 98(5): 1312–20. DOI: 10.1182/blood.V98.5.1312.
4. Schoch C., Haferlach T., Haase D. et al. Patients with de novo acute myeloid leukaemia and complex karyotype aberrations show a poor prognosis despite intensive treatment: A study of 90 patients. Br J Haematol. 2001; 112(1): 118–26. DOI: 10.1046/j.1365-2141.2001.02511.x.
5. Döhner H., Estey E., Grimwade D. et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017; 129: 424–47. DOI: 10.1182/blood-2016-08-733196.
6. Arber D.A., Orazi A., Hasserjian R. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016; 127(20): 2391–405. DOI: 10.1182/blood-2016-03-643544.
7. Ley T.J., Miller C., Ding L. et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013; 368(22): 2059–74. DOI: 10.1056/NEJMoa1301689.
8. Röllig C., Bornhäuser M., Thiede C. et al. Long-term prognosis of acute myeloid leukemia according to the new genetic risk classification of the European LeukemiaNet recommendations: Evaluation of the proposed reporting system. J Clin Oncol. 2011; 29(20): 2758–65. DOI: 10.1200/JCO.2010.32.8500.
9. Preudhomme C., Sagot C., Boissel N. et al. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: A study from the Acute Leukemia French Association (ALFA). Blood. 2002; 100(8): 2717–23. DOI: 10.1182/blood-2002-03-0990.
10. Fröhling S., Schlenk R.F., Stolze I. et al. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: Prognostic relevance and analysis of cooperating mutations. J Clin Oncol. 2004; 22(4): 624–33. DOI: 10.1200/JCO.2004.06.060.
11. Tang J.L., Hou H.A., Chen C.Y. et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: Prognostic implication and interaction with other gene alterations. Blood. 2009; 114(26): 5352–61. DOI: 10.1182/blood-2009-05-223784.
12. Gaidzik V.I., Bullinger L., Schlenk R.F. et al. RUNX1 mutations in acute myeloid leukemia: Results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011; 29(10): 1364–72. DOI: 10.1200/JCO.2010.30.7926.
13. Mendler J.H., Maharry K., Radmacher M.D. et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and microRNA expression signatures. J Clin Oncol. 2012; 30(25): 3109–18. DOI: 10.1200/JCO.2011.40.6652.
14. Gaidzik V.I., Teleanu V., Papaemmanuil E. et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia. 2016; 30: 2160–8. DOI: 10.1038/leu.2016.126.
15. Haferlach T., Stengel A., Eckstein S. et al. The new provisional WHO entity “RUNX1 mutated AML” shows specific genetics but no prognostic influence of dysplasia. Leukemia. 2016; 30(10): 2109–12. DOI: 10.1038/leu.2016.150.
16. Wouters B.J., Löwenberg B., Erpelinck-Verschueren C.A.J. et al. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009; 113(13): 3088–91. DOI: 10.1182/blood-2008-09-179895.
17. Metzeler K.H., Becker H., Maharry K. et al. ASXL1 mutations identify a highrisk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011; 118(26): 6920–9. DOI: 10.1182/blood-2011-08-368225.
18. Pratcorona M., Abbas S., Sanders M.A. et al. Acquired mutations in ASXL1 in acute myeloid leukemia: Prevalence and prognostic value. Haematologica. 2012; 97(3): 388–92. DOI: 10.3324/haematol.2011.051532.
19. Schnittger S., Eder C., Jeromin S. et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. 2013; 27(1): 82–91. DOI: 10.1038/leu.2012.262.
20. Paschka P., Schlenk R.F., Gaidzik V.I. et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: A study by the German-Austrian acute myeloid leukemia study group. Haematologica. 2015; 100(3): 324–30. DOI: 10.3324/haematol.2014.114157.
21. Haferlach C., Dicker F., Herholz H. et al. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia. 2008; 22(8): 1539–41. DOI: 10.1038/leu.2008.143.
22. Bowen D., Groves M.J., Burnett A.K. et al. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia. 2009; 23(1): 203–6. DOI: 10.1038/leu.2008.173.
23. Rücker F.G., Schlenk R.F., Bullinger L. et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012; 119(9): 2114–21. DOI: 10.1182/blood-2011-08-375758.
24. Devillier R., Mansat-De Mas V., Gelsi-Boyer V. et al. Role of ASXL1 and TP53 mutations in the molecular classification and prognosis of acute myeloid leukemias with myelodysplasiarelated changes. Oncotarget. 2015; 6(10): 8388–96. DOI: 10.18632/oncotarget.3460.
25. Tsai C.H., Hou H.A., Tang J.L. et al. Genetic alterations and their clinical implications in older patients with acute myeloid leukemia. Leukemia. 2016; 30(7): 1485–92. DOI: 10.1038/leu.2016.65.
26. Kottaridis P.D., Gale R.E., Frew M.E. et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: Analysis of 854 patients from the United King. Blood. 2001; 98(6): 1752–9. DOI: 10.1182/blood.V98.6.1752.
27. Thiede C., Steudel C., Mohr B. et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002; 99(12): 4326–35. DOI: 10.1182/blood.V99.12.4326.
28. Schlenk R.F., Breitruck J., Benner A. et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002; 100(13): 4372–80. DOI: 10.1182/blood-2002-05-1440.
29. Gale R.E., Green C., Allen C. et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008; 111(5): 2776–84. DOI: 10.1182/blood-2007-08-109090.
30. Pratcorona M., Brunet S., Nomdedéu J. et al. Favorable outcome of patients with AML harboring a low-allelic burden FLT3-ITD and concomitant NPM1 mutation: relevance to post-remission. Blood. 2013; 121(14): 2734–8. DOI: 10.1182/blood-2012-06-431122.
31. Schlenk R.F., Kayser S., Bullinger L. et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. 2014; 124(23): 3441–9. DOI: 10.1182/blood-2014-05-578070.
32. Linch D.C., Hills R.K., Burnett A.K. et al. Impact of FLT3ITD mutant allele level on relapse risk in intermediate-risk acute myeloid leukemia. Blood. 2014; 124(2): 273–6. DOI: 10.1182/blood-2014-02-554667.
33. Papaemmanuil E., Gerstung M., Bullinger L. et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016; 374(23): 2209–21. DOI: 10.1056/NEJMoa1516192.
34. Metzeler K.H., Herold T., Rothenberg-Thurley M. et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016; 128(5): 686–98. DOI: 10.1182/blood-2016-01-693879.
35. Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38(16): 1–7. DOI: 10.1093/nar/gkq603.
36. Tate J.G., Bamford S., Jubb H.C. et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018; 47(D1): D941–7. DOI: 10.1093/nar/gky1015.
37. Scholl S., Mügge L.-O., Landt O. et al. Rapid screening and sensitive detection of NPM1 (nucleophosmin) exon 12 mutations in acute myeloid leukaemia. Leuk Res. 2007; 31(9): 1205–11. DOI: 10.1016/j.leukres.2006.12.011.
38. Lin L.I., Lin T.C., Chou W.C. et al. A novel fluorescence-based multiplex PCR assay for rapid simultaneous detection of CEBPA mutations and NPM mutations in patients with acute myeloid leukemias. Leukemia. 2006; 20(10): 1899–903. DOI: 10.1038/sj.leu.2404331.
39. Brownstein M.J., Carpten J.D., Smith J.R. Modulation of non-templated nucleotide addition by Taq DNA polymerase: Primer modifications that facilitate genotyping. Biotechniques. 1996; 20(6): 1004–10. DOI: 10.2144/96206st01.
40. Ley T.J., Ding L., Walter M.J. et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010; 363(25): 2424–33. DOI: 10.1056/NEJMoa1005143.
41. Di Nardo C.D., Cortes J.E. Mutations in AML: Prognostic and therapeutic implications. Hematology. 2016; 2016(1): 348–55. DOI: 10.1182/asheducation-2016.1.348.
42. Lindsley R.C., Mar B.G., Mazzola E. et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015; 125(9): 1367–76. DOI: 10.1182/blood-2014-11-610543.
Кашлакова Анастасия Игоревна, клинический ординатор отделения интенсивной высокодозной химиотерапии гемобластозов и депрессий кроветворения с круглосуточным стационаром
125167, Москва
тел.: +7 (495) 612-45-92
Паровичникова Елена Николаевна, доктор медицинских наук, руководитель отдела химиотерапии гемобластозов, депрессий кроветворения и трансплантации костного мозга
125167, Москва
Бидерман Белла Вениаминовна, кандидат биологических наук, старший научный сотрудник лаборатории молекулярной гематологии
125167, Москва
Сидорова Юлия Владимировна, кандидат медицинских наук, старший научный сотрудник лаборатории молекулярной гематологии
125167, Москва
Чабаева Юлия Александровна, кандидат технических наук, заместитель заведующего информационно-аналитическим отделом
125167, Москва
Троицкая Вера Витальевна, кандидат медицинских наук, заместитель генерального директора по лечебной работе, заведующая отделением интенсивной высокодозной химиотерапии гемобластозов и депрессий кроветворения с круглосуточным стационаром
125167, Москва
Лукьянова Ирина Анатольевна, кандидат медицинских наук, врач отделения интенсивной отделения интенсивной высокодозной химиотерапии гемобластозов и депрессий кроветворения с дневным стационаром
125167, Москва
Кохно Алина Владимировна, кандидат медицинских наук, ведущий научный сотрудник отделения интенсивной высокодозной химиотерапии гемобластозов и депрессий кроветворения с круглосуточным стационаром
125167, Москва
Соколов Андрей Николаевич, кандидат медицинских наук, старший научный сотрудник отделения интенсивной высокодозной химиотерапии гемобластозов и депрессий кроветворения с круглосуточным стационаром
125167, Москва
Судариков Андрей Борисович, доктор биологических наук, заведующий лабораторией молекулярной гематологии
125167, Москва
Обухова Татьяна Никифоровна, кандидат медицинских наук, заведующая лабораторией кариологии
125167, Москва
Савченко Валерий Григорьевич, доктор медицинских наук, профессор, академик РАН, главный внештатный специалист-гематолог МЗ РФ, генеральный директор
125167, Москва
Кашлакова А.И., Паровичникова Е.Н., Бидерман Б.В., Сидорова Ю.В., Чабаева Ю.А., Троицкая В.В., Лукьянова И.А., Кохно А.В., Соколов А.Н., Судариков А.Б., Обухова Т.Н., Савченко В.Г. Определение молекулярно-генетического профиля у взрослых больных острыми миелоидными лейкозами методом секвенирования нового поколения. Гематология и трансфузиология. 2020;65(4):444-459. https://doi.org/10.35754/0234-5730-2020-65-4-444-459
Kashlakova A.I., Parovichnikova E.N., Biderman B.V., Sidorova Y.V., Chabaeva Y.A., Troitskaya V.V., Lukianova I.A., Kokhno A.V., Sokolov A.N., Sudarikov A.B., Obukhova T.N., Savchenko V.G. Next-generation sequencing-based molecular genetic profiling in adults with acute myeloid leukaemia. Russian journal of hematology and transfusiology. 2020;65(4):444-459. (In Russ.) https://doi.org/10.35754/0234-5730-2020-65-4-444-459
![]()
125167, Москва, Новый Зыковский проезд, 4
ФГБУ «НМИЦ гематологии» Минздрава России
тел.: 8-926-816-3887
e-mail: o.levchenko@htjournal.ru
 Группа благоприятного риска:
Группа благоприятного риска:





































