Введение
Множественная миелома (ММ) представляет собой B-клеточную опухоль с дифференцировкой малигнизированных клеток-предшественников до плазматических клеток. Заболеваемость ММ составляет 1 % от всех случаев опухолевых заболеваний [1]. Благодаря широкому использованию ингибиторов протеасом и иммуномодулирующих препаратов, а также высокодозной химиотерапии (ВХТ) с трансплантацией аутологичных гемопоэтических стволовых клеток (ауто-ТГСК), медиана общей выживаемости больных увеличилась до 5–7 лет, однако прогрессия болезни считается неизбежной [1],[2]. Развитие ММ ассоциировано с генетическими изменениями на уровне клеток, приводящими к нарушениям взаимодействий между опухолью, микроокружением и иммунной системой. Одним из проявлений такого взаимодействия является развитие Т-клеточного истощения (T-cell exhaustion), которое фенотипически характеризуется стабильной экспрессией ингибиторных сигнальных «чек-поинт» молекул (ИСМ), таких как PD-1, LAG-3, TIM-3, а функционально приводит к уменьшению секреции провоспалительных цитокинов (интерлейкина-2, интерферона-γ (ИФН-γ) фактора некроза опухоли альфа), выраженному снижению цитотоксической функции и пролиферативного потенциала [3], что препятствует развитию полноценного противоопухолевого иммунного ответа. В то же время экспрессия одной или нескольких ИСМ не является показателем Т-клеточного истощения. ИСМ экспрессируются активированными Т-клетками с сохранной пролиферативной и цитотоксической функциями в норме и при патологии [4],[5].
Увеличение количества Т-клеток, экспрессирующих ИСМ — PD-1, TIM-3, LAG-3 и др., выявлено при классической лимфоме Ходжкина (ЛХ), некоторых неходжкинских лимфомах (НХЛ) и ММ [6],[7],[8]. Использование препаратов моноклональных антител, блокирующих ИСМ, приводит к увеличению выживаемости при резистентном течении ЛХ [9],[10]. Результаты таргетной анти-PD-1 терапии при рецидивирующих/резистентных НХЛ и ММ в настоящее время очень скромны [11],[12].
ВХТ с ауто-ТГСК остается стандартом лечения больных ММ, не имеющих противопоказаний. Проведение ВХТ приводит к деплеции опухолевого клона и к длительной лимфопении; последняя индуцирует периферическую экспансию зрелых лимфоцитов, реинфузированных в составе аутологичного продукта афереза совместно с гемопоэтическими стволовыми клетками (ГСК). Таргетная терапия, направленная против ИСМ, представляется перспективным методом усиления противоопухолевого иммунного ответа в посттрансплантационном периоде [13]. Динамика восстановления Т-клеток, экспрессирующих ИСМ, после ауто-ТГСК, в том числе при ММ, недостаточно изучена, а имеющиеся данные противоречивы. Увеличение экспрессии PD-1 описано в последние два года в моделях трансплантации гемопоэтических стволовых клеток, выполненных на мышах [14],[15], у больных после ауто-ТГСК [14], у больных гемобластозами после трансплантации аллогенных гемопоэтических стволовых клеток [16] и больных системной склеродермией после ауто-ТГСК [17]. У больных ММ не было ранее выявлено различий в содержании ИСМ до и после ВХТ с ауто-ТГСК [18].
Цель настоящей работы — изучить динамику восстановления Т-клеток, экспрессирующих PD-1 и TIM-3, у больных ММ в условиях длительной лимфопении после ВХТ с ауто-ТГСК, а также оценить их функциональные свойства.
Материал и методы
В исследование включены 40 больных ММ, которым в период с марта 2018 г. по январь 2020 г. были проведены ВХТ и ауто-ТГСК на базе ФГБНУ «Научноисследовательский институт фундаментальной и клинической иммунологии» (табл. 1).
Режим мобилизации включал инфузию циклофосфамида (2–4 г/м 2) с последующим введением гранулоцитарного колониестимулирующего фактора (5 мкг/кг/день) в течение 4–5 дней до достижения в периферической крови (ПК) концентрации 10 4 CD34+ CD45+ клеток/мл. Процедуру афереза выполняли на сепараторах клеток крови ASTEC 204 (Fresenius, Германия) и Spectra LRS 07 (COBE) до получения ≧ 2,0 × 10 6 CD34+ CD45+ клеток/кг (1–2 процедуры афереза). Медиана реинфузированных CD34+ CD45+ ГСК составляла 4,55 × 10 6/кг (3,30–5,80 × 10 6/кг). ВХТ перед ауто-ТГСК проводили через 2–6 мес. после сепарации ГСК, в этом промежутке всем больным проводили поддерживающую терапию бортезомибом, при рецидиве ММ использовали программы с леналидомидом (VRD, RD, RCD, монотерапия леналидомидом) и другие комбинации цитостатических препаратов (DCEP, BBD). Режим кондиционирования — мелфалан 140–200 мг/м 2, дозу рассчитывали с учетом сопутствующих заболеваний. Всем больным после ауто-ТГСК проводили поддерживающую терапию бортезомибом или леналидомидом.
Взятия образцов крови, костного мозга (КМ) и все иммунологические исследования проводили после получения письменного информированного согласия больного. Содержание субпопуляций Т-клеток, экспрессирующих PD-1 и TIM-3, оценивали в ПК и КМ больных перед началом кондиционирования (n = 25), после ауто-ТГСК в день выхода из лейкопении (лейкоциты > 1 × 10 9/л, в среднем на 14-й день, n = 28, только ПК) и через 6 мес. (n = 21, ПК и КМ).
Лизис эритроцитов проводили раствором VersaLyse («Beckman Coulter», Франция) в соответствии с инструкцией. Методом проточной цитометрии оценивали относительное содержание CD8+ PD-1+ , CD8+ TIM-3+ , CD8+ PD-1+ TIM-3+ , CD4+ PD-1+ , CD4+ TIM-3+ , CD4+ PD-1+ TIM-3+ T-клеток ПК и КМ, используя анти-CD8 (FITC), анти-CD4 (FITC, PerCP), анти-PD-1 (APC), анти-TIM-3 (PE, PerCP/Cy 5.5) моноклональные антитела («BD Biosciences», США). У 15 больных в отдельных пробах оценили внутриклеточную экспрессию маркера пролиферации Ki-67 (BD Pharmingen™ PE Mouse Anti-Ki-67 Set, «BD Biosciences») среди PD-1-и TIM-3-позитивных и негативных Т-клеток. С целью более щадящей обработки [19] использовали набор растворов для фиксации/пермеабилизации Transcription Factor Buffer Set («BD Biosciences») в соответствии с инструкцией. У 13 больных в субпопуляции CD8+ Т-лимфоцитов исследовали внутриклеточную продукцию гранзима В среди PD-1 и TIM-3 позитивных и негативных клеток, используя анти-гранзим В моноклональные антитела (PE, «BD Biosciences»). Для исследования внутриклеточной продукции ИФН-γ выделяли мононуклеарные клетки (МНК) ПК стандартным методом центрифугирования цельной гепаринизированной венозной крови в градиенте плотности фиколла-верографина (ρ = 1,078). У 11 больных в CD8+ и CD4+ Т-лимфоцитах оценили внутриклеточную продукцию ИФН-γ среди PD-1 и TIM-3 позитивных и негативных клеток, используя анти-ИФН-γ моно-клональные антитела (PE, «BD Biosciences»), после 5-часовой активации 10 6 МНК в культуральной среде RPMI-1640 («БиоЛот», Санкт-Петербург) с форболмиристат ацетатом («Sigma», Германия; конечная концентрация 10 нг/мл суспензии клеток), иономицином («Sigma»; конечная концентрация 10 мкг/мл суспензии клеток) при температуре 37 °C в CO2 -инкубаторе (5 % СО2 ); после первого часа активации в суспензию клеток одновременно добавляли ингибиторы транспорта белков BD GolgiStop™, содержащий монэнзин, и BD GolgiPlug™, содержащий брефелдин А (оба — «BD Biosciences»), в соответствии с инструкцией. Исследование каждого внутриклеточного маркера проводили в отдельных пробах, по отдельности для субпопуляций CD4+ и CD8+ Т-клеток. Фиксацию и пермеабилизацию МНК для оценки внутриклеточной экспрессии Ki-67 и продукции гранзима В и ИФН-γ проводили после инкубации клеток с моноклональными антителами против поверхностных антигенов; использовали набор растворов для фиксации/пермеабилизации Transcription Factor Buffer Set («BD Biosciences»).
Исследование проводили по общепринятой методике с использованием параметров прямого и бокового светорассеяния и флуоресценции по каналам FL-1 (FITC), FL-2 (РЕ), FL-3 (РerCP, PerCP/Cy 5.5, PE-Cy 5), FL-4 (APC) («BD FACSCalibur», «CellQuest Software», США). Анализ проводили после накопления не менее 30 000 событий в регионе CD8+ Т-клеток.
Абсолютное содержание исследуемых субпопуляций клеток рассчитывали как:
субпопуляция Т−клеток, % от лимфоцитов × абсолютное содержание лимфоцитов,×109 /л /100
Абсолютное количество лимфоцитов рассчитывали из результатов общего анализа крови с лейкоцитарной формулой. Общий анализ крови оценивали на автоматическом гематологическом анализаторе Hema-Screen 18 («Hospitex Diagnostics», Италия).
Статистический анализ. Статистическую обработку данных проводили с помощью пакета программ Statistica 6.0 (StatSoft Inc., США) и GraphPad Prism 5 (GraphPad Software, Inc., США). Для оценки значимости различий между двумя независимыми группами использовали U-критерий Манна — Уитни. Для оценки значимости различий зависимых выборок использовали критерий Вилкоксона для парных выборок. Данные представлены в виде медианы (Ме) и интерквартильного интервала (МКИ), если не указано другого. Различия считали статистически значимыми при уровне значимости p < 0,05 (двустороннем).
Результаты
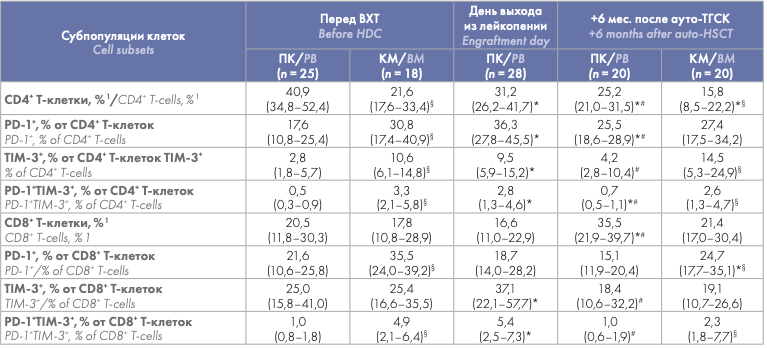
Проведена сравнительная оценка содержания CD4+ и CD8+ Т-клеток, экспрессировавших PD-1 и TIM-3, в циркуляции и КМ до ауто-ТГСК, на момент выхода из лейкопении (только ПК) и через 6 мес. после ауто-ТГСК. Относительное количество большинства субпопуляций циркулировавших Т-клеток, экспрессировавших PD-1 и TIM-3, было значимо меньше соответствующих значений в КМ как до ВХТ, так и через 6 мес. после ауто-ТГСК. Не выявлено различий между содержанием в ПК и КМ только для TIM-3+ субпопуляции CD8+ Т-клеток (на обеих точках) и для PD-1+ CD4+ Т-клеток через 6 мес. после ауто-ТГСК (табл. 2). Выявлено транзиторное увеличение содержания PD-1 и TIM-3-позитивных субпопуляций в циркулирующих Т-клетках в раннем посттрансплантационном периоде. Процентное содержание большинства исследуемых субпопуляций Т- клеток (кроме PD-1+ CD8+ Т-клеток) на день выхода из лейкопении (в среднем, 14-е сутки после ауто-ТГСК) было значимо больше как исходных показателей, так и их значений через 6 мес. наблюдения. Содержание TIM-3+ CD4+ Т-клеток и PD-1+ и TIM-3+ субпопуляций CD8+ Т-клеток через 6 мес. после ауто-ТГСК не отличалось от соответствующих показателей перед ВХТ, а содержание PD-1+ и PD-1+ TIM-3+ субпопуляций CD4+ Т-клеток оставалось более высоким (табл. 2).
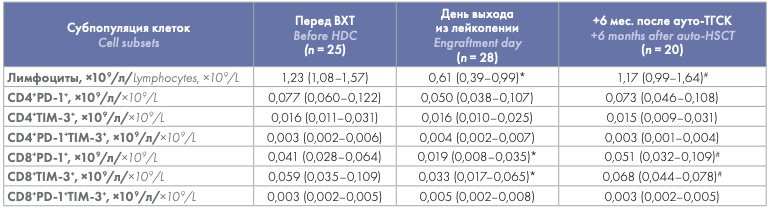
С учетом меньшего абсолютного количества лимфоцитов у больных на день выхода из лейкопении и последующего их восстановления, абсолютные значения CD4+ PD-1+ , CD4+ TIM-3+ , CD4+ PD-1+ TIM-3+ и CD8+ PD-1+ TIM-3+ Т-клеток ПК в течение периода наблюдения после ауто-ТГСК значимо не отличались от предтрансплантационных значений. Абсолютные показатели CD8+ PD-1+ и CD8+ TIM-3+ Т-клеток были значимо снижены на день выхода из лейкопении по сравнению со значениями до ауто-ТГСК, однако через 6 мес. наблюдения достигали исходных значений (табл. 3).
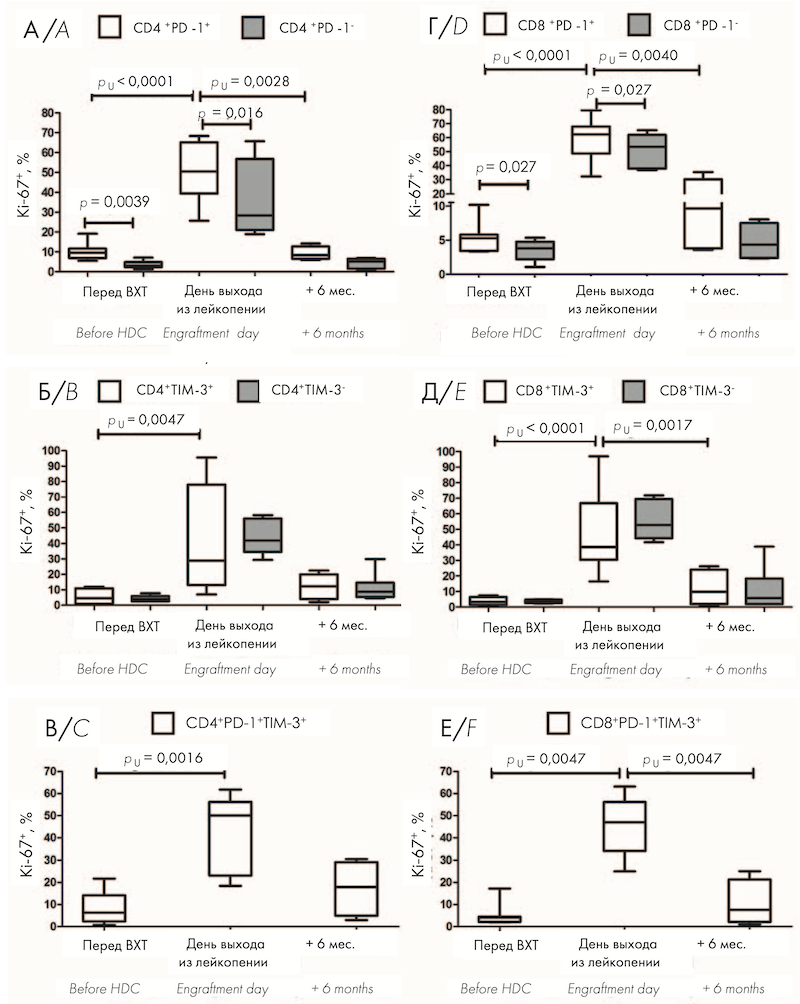
Учитывая высокое относительное содержание циркулирующих Т-клеток, экспрессирующих PD-1 и TIM-3 в раннем посттрансплантационном периоде, проведена оценка внутриклеточной экспрессии маркера пролиферации Ki-67. Перед ауто-ТГСК относительное количество пролиферирующих Ki-67+ клеток было значимо больше среди PD-1-позитивных CD4+ и CD8+ Т-клеток по сравнению с PD-1-негативными клетками (рис. 1, А, Г). TIM-3+ Т-клетки не отличались по содержанию Ki-67 от TIM-3- клеток (рис. 1, Б, Д). В раннем посттрансплантационном периоде — на день выхода из лейкопении после ауто-ТГСК — отмечено выраженное увеличение пролиферативной активности всех исследуемых субпопуляций клеток по сравнению с предтрансплантационными показателями (рис. 1, А–Е), причем доля Ki-67-позитивных клеток среди PD-1+ T-лимфоцитов оставалась значимо больше по сравнению с PD-1- лимфоцитами (рис. 1, А, Г). Через 6 мес. после ауто-ТГСК относительное количество Ki-67-позитивных клеток среди всех исследуемых субпопуляций не отличалось от значений перед кондиционированием (рис. 1, А–Е), при этом доля активно пролиферирующих CD4+ PD-1+ , CD8+ PD-1+ , CD8+ TIM-3+ , CD8+ PD-1+ TIM-3+ Т-клеток была значимо меньше соответствующих показателей на день выхода из лейкопении (рис. 1, А, Г–Е). На рисунке 1 представлено относительное количество Ki-67+ клеток среди PD-1-позитивных и негативных CD4+ (А) и CD8+ (Г) T-клеток, среди TIM-3-позитивных и негативных CD4+ (Б) и CD8+ (Д) T-клеток, а также в субпопуляциях CD4+ PD-1+ TIM-3+ (В) и CD8+ PD-1+ TIM-3+ (Е) T-клеток в ПК больных ММ перед кондиционированием (n = 9) и после ауто-ТГСК: на день выхода из лейкопении (n = 10) и через 6 мес. (n = 5).
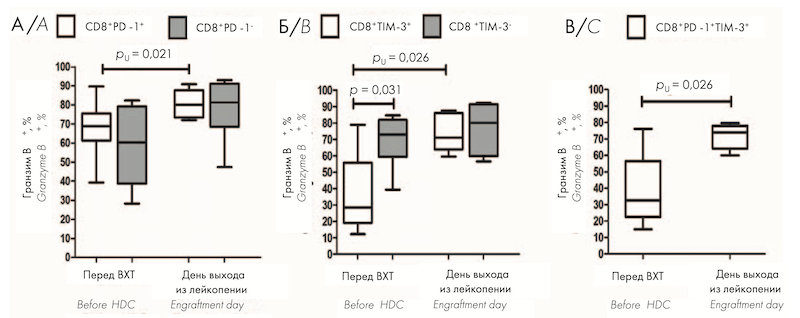
Т-клеткам, находящимся в состоянии истощения, не свойственна высокая пролиферативная активность. Для изучения других функциональных свойств Т-клеток, экспрессирующих ингибиторные ИСМ PD-1 и TIM-3 в раннем посттрансплантационном периоде, проведена оценка их цитотоксического и цитокин-продуцирующего потенциала перед кондиционированием и на день выхода из лейкопении после ауто-ТГСК. Перед ВХТ CD8+ PD-1+ Т-клетки не отличались по содержанию гранзима В от PD-1– субпопуляции (рис. 2, А); относительное количество гранзим В-продуцирующих клеток было значимо меньше среди CD8+ TIM-3+ Т-клеток по сравнению с CD8+ TIM-3– субпопуляцией (рис. 2, Б). На день выхода из лейкопении после ауто-ТГСК продукция гранзима В CD8+ PD-1+ , CD8+ TIM-3+ и CD8+ PD-1+ TIM-3+ Т-клетками была значимо больше по сравнению с показателями перед ауто-ТГСК (рис. 2, А–В); не выявлено различий в содержании гранзима В между PD-1-и TIM-3-позитивными и негативными популяциями CD8+ T-клеток (рис. 2, А, Б). На рисунке 2 представлено относительное количество гранзим В+ клеток среди PD-1-позитивных и негативных (А) и среди TIM-3-позитивных и негативных (Б) CD8+ T-клеток, а также в субпопуляции CD8+ PD-1+ TIM-3+ (В) T-клеток в ПК больных ММ перед кондиционированием (n = 10) и на день выхода из лейкопении (n = 8) после ауто-ТГСК.
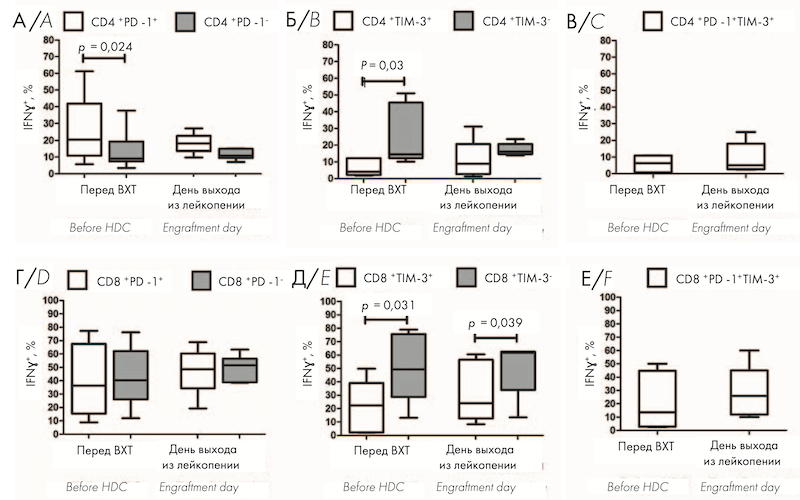
Перед ауто-ТГСК относительное количество ИФН-γ-продуцирующих клеток было значимо больше среди CD4+ PD-1+ Т-клеток по сравнению с CD4+ PD-1– клетками (рис. 3, A); CD8+ PD-1+ Т-клетки не отличались по содержанию ИФН-γ от PD-1– клеток (рис. 3, Г). Продукция ИФН-γ была значимо меньше в CD8+ TIM-3+ и CD4+ TIM-3+ клетках по сравнению с Т-клетками, не экспрессирующими TIM-3 (рис. 3, Б, Д). На день выхода из лейкопении после аутоТГСК не выявлено значимых изменений продукции ИФН-γ среди всех исследуемых субпопуляций клеток по сравнению с предтрансплантационными показателями (рис. 3, А–Е); при этом доля ИФН-γ -позитивных клеток среди CD8+ TIM-3+ T-лимфоцитов оставалась значимо большей по сравнению с TIM-3-негативными Т-лимфоцитами (рис. 3, Д). На рисунке 3 представлено относительное количество ИФН-γ+ клеток среди PD-1-позитивных и негативных CD4+ (А) и CD8+ (Г) T-клеток, среди TIM-3-позитивных и негативных CD4+ (Б) и CD8+ (Д) T-клеток, а также в субпопуляциях CD4+ PD-1+ TIM-3+ (В) и CD8+ PD-1+ TIM-3+ (Е) T-клеток в ПК больных ММ перед кондиционированием (n = 9) и на день выхода из лейкопении (n = 6) после ауто-ТГСК.
Обсуждение
Теоретическим обоснованием использования моноклональных антител против ИСМ или их лигандов является увеличение экспрессии этих молекул на эффекторных Т-клетках при онкологических заболеваниях. Передача сигнала через ИСМ — PD-1, TIM-3, LAG-3 и др. — в норме играет регулирующую роль, ограничивая интенсивность иммунного ответа. Также экспрессия ИСМ ассоциирована с Т-клеточным истощением, состоянием дисфункции Т-клеток памяти, возникающим в условиях постоянной антигенной стимуляции. Повышенная экспрессия соответствующих лигандов ИСМ, например, PD-L1 и PD-L2 для PD-1, малигнизированными клетками и/или их микроокружением является одним из путей «ухода» опухоли из-под иммунного надзора [3]. Моноклональные антитела (анти-PD-1, анти-PDL1, анти-CTLA-4) конкурентно блокируют взаимодействия между лигандом и рецептором, предотвращая ослабление противоопухолевого иммунного ответа. Попытки использования терапевтических моноклональных анти-PD-1 антител при различных вариантах течения и этапах терапии ММ не привели к значимым клиническим результатам, хотя отмечено большое количество иммуноопосредованных нежелательных реакций. Однако исследования в этом направлении продолжаются [20]. Настоящая работа посвящена количественной характеристике и изучению функциональных свойств циркулирующих Т-клеток, экспрессирующих PD-1 и TIM-3, у больных ММ после ВХТ с ауто-ТГСК.
Относительное количество большинства субпопуляций Т-клеток, экспрессирующих PD-1 и TIM-3 ИСМ, было больше в образцах КМ по сравнению с ПК (не выявлено различий для CD8+ TIM-3+ Т-клеток) как перед ВХТ, так и через 6 мес. после ауто-ТГСК. С учетом преимущественного поражения опухолевым процессом КМ, описанное распределение дисфункциональных Т-клеток представляется закономерным, однако ранее опубликованные данные противоречивы. С. Zelle-Rieser и соавт. [6] на небольшой выборке (n = 7–12) не обнаружили различий между содержанием PD-1+ клеток в КМ и ПК. J. Tan и соавт. [21] показали большее содержание CD8+ PD-1+ субпопуляции в КМ по сравнению с ПК, в то время как содержание CD4+ PD-1+ Т-клеток и TIM-3+ T-клеток (включая PD-1+ TIM-3+ субпопуляции) в КМ значимо не отличалось от таковых в ПК 10 больных ММ при проведении лечения.
В настоящей работе показано выраженное увеличение содержания большинства PD-1-и TIM-3-экспрессирующих субпопуляций Т-клеток (не выявлено различий для CD8+ PD-1+ Т-клеток) в раннем посттрансплантационном периоде — на день выхода из лейкопении после ауто-ТГСК, — которое не зависело от относительного содержания CD4+ и CD8+ Т-клеток. Через 6 мес. доля PD-1-и TIM-3-положительных Т-клеток была значимо меньше, чем при восстановлении, либо достигала предтрансплантационных значений, либо несколько превышала их. В работах N. Marshall и соавт. [14] и F. Simonetta и соавт. [16], исследовавших уровень экспрессии PD-1 после ауто-ТГСК и трансплантации аллогенных гемопоэтических стволовых клеток, соответственно, также было показано неожиданное увеличение экспрессии PD-1 и относительного содержания PD-1+ Т-клеток после выхода из лейкопении. Ранее D. J. Chung и соавт. [18] описали сохранение экспрессии ИСМ, в том числе PD-1 и TIM-3, Т-клетками ПК больных ММ через 3 мес. после ауто-ТГСК на предтрансплантационном уровне, что также согласуется с нашими данными о восстановлении на 6 мес. после ауто-ТГСК.
Учитывая высокое относительное содержание циркулирующих Т-клеток, экспрессирующих PD-1 и TIM-3 в раннем посттрансплантационном периоде, была проведена оценка внутриклеточной экспрессии маркера пролиферации Ki-67 в исследуемых субпопуляциях до и после ауто-ТГСК. Выявлено значительное увеличение доли активно пролиферирующих PD-1- и TIM-3-положительных Т-клеток на день выхода из лейкопении по сравнению с показателями до ВХТ, при этом процент Ki-67-позитивных клеток среди PD-1+ T-лимфоцитов был значимо больше по сравнению с PD-1- Т-лимфоцитами как перед кондиционированием, так и на день выхода из лейкопении. Через 6 мес. после ауто-ТГСК интенсивность пролиферации уменьшалась до исходных значений.
Т-клетки в состоянии истощения помимо сниженного пролиферативного потенциала характеризуются уменьшением цитотоксической и цитокин-продуцирующей активности. Для оценки этих свойств PD-1-и TIM-3-экспрессирующих Т-клеток исследована продукция гранзима В и ИФН-γ до и после ауто-ТГСК. Показано, что значительная часть CD8+ PD-1+ и CD4+ PD-1+ Т-клеток больных ММ перед кондиционированием обладала выраженным цитотоксическим и цитокин-продуцирующим потенциалом, не отличаясь от PD-1-негативного компартмента, либо превосходя его. В данном случае экспрессию PD-1, вероятно, нужно рассматривать в качестве маркера активированных, либо «ранних дисфункциональных», но не терминально истощенных Т-клеток. F. M. Schnorfeil и соавт. [5] ранее описали сохранную функциональную активность PD-1+ CD4+ и CD8+ Т-клеток при остром миелоидном лейкозе, сделав заключение об отсутствии значимой клинической роли Т-клеточного истощения в развитии болезни. Сохранение либо даже усиление цитотоксических, ИФН-γ-продуцирующих и пролиферативных свойств PD-1-экспрессирующими Т-клетками на ранних этапах Т-клеточной дисфункции/истощения было недавно описано при меланоме [22],[23], немелкоклеточном раке легкого [24] и гепатоцеллюлярной карциноме [25].
Функциональная активность CD8+ TIM-3+ и CD4+ TIM-3+ Т-клеток, напротив, была значимо снижена по сравнению с TIM-3-негативными Т-клетками. Можно предположить, что, несмотря на описанную в экспериментах in vitro возможность экспрессии на активированных Т-клетках [4],[26], молекула TIM-3 более ассоциирована с состоянием Т-клеточного истощения, чем PD-1. Ранее Z. Li и соавт. [27] описали более выраженные нарушения продукции CD8+ TIM-3+ PD-1– Т-клетками цитокинов, гранзима В и перфорина по сравнению с CD8+ PD-1+ TIM-3– клетками у больных злокачественной шванномой. Подобных данных для гематологических заболеваний ранее опубликовано не было, и регулирующая роль этого рецептора при ММ требует дальнейшего изучения.
Низкие уровни Ki-67-экспрессирующих гранзим В- и ИФН-γ-продуцирующих клеток в субпопуляциях TIM-3+ PD-1+ Т-клеток согласуются с данными литературы о совместной экспрессии двух и более ИСМ как маркера Т-клеточного истощения [3], [22],[23],[24],[25],[28].
Относительное количество гранзим В-продуцирующих клеток среди CD8+ PD-1+ , CD8+ TIM-3+ и CD8+ PD-1+ TIM-3+ Т-клеток было значимо увеличено после ауто-ТГСК на день выхода из лейкопении по сравнению с показателями до ВХТ, при этом цитотоксический потенциал CD8+ TIM-3+ клеток достигал значений TIM-3-негативного пула. Доля ИФН-γпродуцирующих клеток среди всех исследованных PD-1+ и TIM-3+ субпопуляций Т-клеток после аутоТГСК соответствовала уровню до ВХТ.
Интерес представляет описанное в настоящей работе значительное увеличение активно делящихся PD-1-и TIM-3-экспрессирующих Т-клеток в условиях лимфопении после ВХТ с ауто-ТГСК, которое могло быть инициировано гомеостатической пролиферацией зрелых лимфоцитов. В настоящее время отсутствуют данные о влиянии гомеостатических цитокинов интерлейкинов -2, -7 и -15 на экспрессию ИСМ и/или экспансию и свойства дисфункциональных/истощенных Т-клеток. Состояние Т-клеточного истощения возникает вследствие длительной антигенной стимуляции через Т-клеточный рецептор (ТКР), в то время как гомеостатическая пролиферация — цитокинопосредованный ТКР-независимый процесс [29]. Учитывая растущий интерес к таргетной терапии, направленной на блокаду ингибиторных чек-поинт рецепторов и их лигандов, при ММ, необходимы дальнейшие исследования экспансии Т-клеток в условиях лимфопении, как фактора, способного изменять количественные и функциональные характеристики клеток-мишеней.
Исходя из полученных данных, можно заключить, что выявленное более высокое содержание циркулирующих PD-1-позитивных Т-клеток у больных ММ ассоциировано с субпопуляциями, не находящимися в состоянии терминального Т-клеточного истощения и сохранившими свои функциональные свойства. Относительное количество функционально активных клеток было более низким среди CD8+ TIM-3+ и CD4+ TIM-3+ Т-клеток, а также в субпопуляциях, ко-экспрессирующих PD-1 и TIM-3. Для идентификации состояния Т-клеточного истощения необходима оценка субпопуляций Т-клеток, ко-экспрессирующих PD-1, TIM-3 и другие ИСМ, и/или исследование их функциональных свойств. Относительное количество Т-клеток, экспрессирующих PD-1 и TIM-3, значимо увеличивается после ВХТ с ауто-ТГСК. В раннем посттрансплантационном периоде также значительно увеличиваются пролиферативная активность PD-1+ и TIM-3+ CD4+ и CD8+ Т-клеток и цитотоксический потенциал PD-1+ и TIM-3+ CD8+ Т-клеток по сравнению с показателями до ауто-ТГСК.
Список литературы
1. Kazandjian D. Multiple myeloma epidemiology and survival: A unique malignancy. Semin Oncol. 2016; 43(6): 676–81. DOI: 10.1053/j.seminoncol.2016.11.004.
2. Kumar S.K., Rajkumar S.V., Dispenzieri A., et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008; 111(5): 2516–20. DOI: 10.1182/blood-2007-10-116129.
3. Wherry E.J., Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015; 15(8): 486–99. DOI: 10.1038/nri3862.
4. Sabins N.C., Harman B.C., Barone L.R., et al. Differential expression of immune checkpoint modulators on in vitro primed CD4+ and CD8+ T cells. Front Immunol. 2016; 7: 221. DOI: 10.3389/fi mmu.2016.00221.
5. Schnorfeil F.M., Lichtenegger F.S., Emmerig K., et al. T cells are functionally not impaired in AML: Increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J Hematol Oncol. 2015; 8: 93. DOI: 10.1186/s13045-015-0189-2.
6. Zelle-Rieser C., Thangavadivel S., Biedermann R., et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol. 2016; 9(1): 116. DOI: 10.1186/s13045-016-0345-3.
7. Pianko M.J., Liu Y., Bagchi S., Lesokhin A.M. Immune checkpoint blockade for hematologic malignancies: A review. Stem Cell Investig. 2017; 4: 32. DOI: 10.21037/sci.2017.03.04.
8. Görgün G., Samur M.K., Cowens K.B., et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res. 2015; 21(20): 4607–18. DOI: 10.1158/1078-0432.CCR-15-0200.
9. Armand P., Engert A., Younes A., et al. Nivolumab for relapsed/refractory classic Hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: Extended follow-up of the multicohort single-arm phase II CheckMate 205 trial. J Clin Oncol. 2018; 36(14): 1428–39. DOI: 10.1200/JCO.2017.76.0793.
10. Chen R., Zinzani P.L., Fanale M.A., et al. Phase II study of the effi cacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol. 2017; 35(19): 2125–32. DOI: 10.1200/JCO.2016.72.1316.
11. Lesokhin A.M., Ansell S.M., Armand P., et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: Preliminary results of a phase Ib study. J Clin Oncol. 2016; 34(23): 2698–704. DOI: 10.1200/JCO.2015.65.9789.
12. Paiva B., Azpilikueta A., Puig N., et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia. 2015; 29(10): 2110–3. DOI: 10.1038/leu.2015.79.
13. Benson D.M. Jr. Checkpoint inhibition in myeloma. Hematology Am Soc Hematol Educ Program. 2016; 2016(1): 528–33. DOI: 10.1182/asheducation-2016.1.528.
14. Marshall N., Hutchinson K., Marron T.U., et al. Antitumor T-cell homeostatic activation is uncoupled from homeostatic inhibition by checkpoint blockade. Cancer Discov. 2019; 9(11): 1520–37. DOI: 10.1158/2159-8290.CD-19-0391.
15. Minnie S.A., Kuns R.D., Gartlan K.H., et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood. 2018; 132(16): 1675–88. DOI: 10.1182/blood-2018-01-825240.
16. Simonetta F., Pradier A., Bosshard C., et al. Dynamics of expression of programmed cell death protein-1 (PD-1) on T cells after allogeneic hematopoietic stem cell transplantation. Front Immunol. 2019; 10:1034. DOI: 10.3389/fi mmu.2019.01034.
17. Arruda L.C.M., Lima-Júnior J.R., Clave E., et al. Homeostatic proliferation leads to telomere attrition and increased PD-1 expression after autologous hematopoietic SCT for systemic sclerosis. Bone Marrow Transplant. 2018; 53(10): 1319–27. DOI: 10.1038/s41409-018-0162-0.
18. Chung D.J., Pronschinske K.B., Shyer J.A., et al. T-cell exhaustion in multiple myeloma relapse after autotransplant: optimal timing of immunotherapy. Cancer Immunol Res. 2016; 4(1): 61–71. DOI: 10.1158/2326-6066.CIR-15-0055.
19. Sun Y., Yang K., Bridal T., Ehrhardt A.G. Robust Ki67 detection in human blood by fl ow cytometry for clinical studies. Bioanalysis. 2016; 8(23): 2399–413. DOI: 10.4155/bio-2016-0194.
20. Jelinek T., Paiva B., Hajek R. Update on PD-1/PD-L1 inhibitors in multiple myeloma. Front Immunol. 2018; 9: 2431. DOI: 10.3389/fi mmu.2018.02431.
21. Tan J., Chen S., Huang J., et al. Increased exhausted CD8+ T cells with programmed death-1, T-cell immunoglobulin and mucin-domain-containing-3 phenotype in patients with multiple myeloma. Asia Pac J Clin Oncol. 2018; 14(5): e266–74. DOI: 10.1111/ajco.13033.
22. Li H., van der Leun A.M., Yofe I., et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2020; 181(3): 747. DOI: 10.1016/j.cell.2020.04.017.
23. Miller B.C., Sen D.R., Al Abosy R., et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019; 20(3): 326–36. DOI: 10.1038/s41590-019-0312-6.
24. Thommen D.S., Koelzer V.H., Herzig P., et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-smallcell lung cancer treated with PD-1 blockade. Nat Med. 2018; 24: 994–1004. DOI: 10.1038/s41591-018-0057-z.
25. Ma J., Zheng B., Goswami S., et al. PD1Hi CD8+ T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J Immunother Сancer. 2019; 7(1): 331. DOI: 10.1186/s40425-019-0814-7.
26. Hastings W.D., Anderson D.E., Kassam N., et al. TIM-3 is expressed on activated human CD4+ T cells and regulates Th1 and Th17 cytokines. Eur J Immunol. 2009; 39(9): 2492–501. DOI: 10.1002/eji.200939274.
27. Li Z., Liu X., Guo R., Wang P. TIM-3 plays a more important role than PD-1 in the functional impairments of cytotoxic T cells of malignant Schwannomas. Tumour Biol. 2017; 39(5): 1010428317698352. DOI: 10.1177/1010428317698352.
28. Sakuishi K., Apetoh L., Sullivan J.M., et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010; 207(10): 2187–94. DOI: 10.1084/jem.20100643.
29. Goldrath A.W., Luckey C.J., Park R., et al. The molecular program induced in T cells undergoing homeostatic proliferation. Proc Natl Acad Sci USA. 2004;101(48): 16885–90. DOI: 10.1073/pnas.0407417101.


 Е. В. Баторов,
Е. В. Баторов,